способ определения микроколичеств общего хрома в воде. Определение в воде хрома
Способ определения микроколичеств общего хрома в воде
Изобретение относится к области аналитической химии, а именно к способу определения общего хрома (CrIII и CrVI) в воде рыбохозяйственных водоемов с нормой по содержанию хрома не более 0,001 мг/дм3. Способ включает предварительное концентрирование хрома путем выпаривания и обработку, позволяющую устранить мешающее влияние примесей фотометрическому определению хрома по реакции с дифенилкарбазидом.
Изобретение относится к области аналитической химии, а именно к способу определения общего содержания хрома (CrIII и CrVI) в воде рыбохозяйственных водоемов с содержанием хрома в пределах 0,001 до 0,01 мг/дм3.
Известен способ определения хрома методом пламенной атомно-абсорбционной спектрометрии (ПНДФ 14.1:2:22-95. Количественный химический анализ вод. Методика выполнения измерений массовой концентрации железа, кадмия, свинца, цинка и хрома в пробах природных и сточных вод методом пламенной атомно-абсорбционной спектрометрии), чувствительность метода 0,02 мг/дм3. Недостатком способа является его недостаточная чувствительность для анализа вод с нормой по содержанию хрома "не более 0,001 мг/дм3", к каким могут быть отнесены пробы воды с показателем pH от 7,0 до 8,5, содержащих восстановители и (или) окрашенные органические вещества, в которых из-за невозможности раздельного определения CrIII и CrVI, определяют только общее содержание хрома, однако, для санитарно-гигиенической оценки качества воды расчеты ведут по наиболее токсичному (ПДК = 0,001 мг/дм3) хрому VI. (Ю.В. Новиков и др., Методы исследования качества воды водоемов, Изд. "Медицина", М. 1990, с. 228). Известен способ фотометрического определения малых количеств суммарного содержания хрома в водах, имеющих щелочную реакцию или содержащих большие количества органических веществ, хлоридов, по которому в пробе, содержащем от 0,001 до 0,05 мг хрома, сначала восстанавливают хром (VI) до хрома (III) сернистой кислотой, затем осаждают хром (III) оксидом магния, отфильтровывают, высушивают, сжигают, растворяют в серной кислоте, окисляют хром (III) до хрома (VI) персульфатом аммония или прокаливанием со смесью карбоната натрия и оксида магния, приливают раствор дифенилкарбазида и фотометрируют. (Ю. Ю. Лурье. Аналитическая химия промышленных сточных вод, Изд. "Химия". М. 1984. с. 155.). Недостатком способа является трудоемкость и высокая погрешность определения, связанная с его многостадийностью, и недостаточная чувствительность (0,02 мг/дм3) для анализа природных вод с нормой по содержанию хрома 0,001 мг/дм3. Известен способ определения хрома в промышленных сточных водах рентгенофлуоресцентным методом (ПНДФ 14.1.43.96. Количественный химический анализ вод. Методика выполнения измерений массовой концентрации ванадия, хрома, марганца, железа, кобальта, никеля, меди, цинка, свинца и висмута в промышленных сточных водах рентгенофлуоресцентным методом), чувствительность метода 0,01 мг/дм3. Недостатком способа является его низкая чувствительность и необходимость дорогостоящего специального оборудования. Известен способ определения общего хрома в воде с помощью атомно-абсорбционной спектрометрии при электротермической атомизации пробы (Г.С. Фомин, Вода. Контроль химической, бактериальной и радиационной безопасности по международным стандартам. Энциклопедический справочник. ИСО 9174, Метод "В",), чувствительность метода 0,005 мг/дм3. Недостатком способа является его неприемлемость для аналитического контроля воды рыбохозяйственных водоемов, для которых предельно допустимая концентрация (ПДК) по содержанию хрома составляет 0,001 мг/дм3. Известен способ определения общего хрома в пробах природной питьевой и сточной воды на анализаторе Флюорат-2, основанный на регистрации свечения хемилюминесценции реакции окисления люминола раствором пероксида водорода, катализируемого солями хрома (III) в щелочной среде (ПНДФ 14.1:2:4. 30-95, Количественный химический анализ вод. Методика выполнения измерений массовой концентрации хрома общего в пробах природной, питьевой и сточной воды на анализаторе жидкости "Флюорат-02"), Минимально определяемая концентрация 0,002 мг/дм3. Недостатком способа является необходимость применения дорогостоящего специального оборудования. Известен способ определения общего хрома в пробах питьевых природных и сточных вод методом инверсионной вольтамперометрии, основанный на адсорбционном концентрировании на поверхности графитового электрода 1,5-дифенилкарбозоната хрома (III), образовавшегося в результате реакции дифенилкарбозона с ионами Cr(VI). Минимально определяемая концентрация хрома 0,001 мг/дм3 (ПНДФ 14.1:2:4.72-96, Количественный химический анализ вод. Методика выполнения измерений массовой концентрации хрома в пробах питьевых, природных и сточных вод методом инверсионной вольтамперометрии.). Недостатком способа является его неприемлемость для анализа вод, содержащих органические загрязнения (содержание органического углерода не должно превышать 0,001 мг/дм3) и необходимость применения специфического оборудования. Наиболее близким по технической сущности и достигаемому результату является способ фотометрического определения общего содержания хрома в природных и сточных водах по реакции хрома (VI) с дифенилкарбазидом, по которому отбирают такой объем исследуемой пробы (разбавленной, сконцентрированной упариванием, минерализованной), в котором содержится (0,001-0,05) мг хрома, доводят объем до 100 см3, нейтрализуют пробу до pH=8 кислотой или щелочью, добавляют 0,3 см3 раствора серной кислоты с концентрацией с(1/2h3SO4)=1 моль/дм3 (1: 2:1000 моль), (5-10) см3 0,1% раствора персульфата аммония (от 0,005 до 0,01 г), кипятят 20-25 мин, фильтруют, упаривают фильтрат до объема 50 см3, нейтрализуют до pH=8, приливают 1 см3 серной кислоты (1:1) и 0,3 см3 концентрированной фосфорной кислоты, доливают раствор до 100 см3 дистиллированной водой, перемешивают, добавляют 2 см3 раствора дифенилкарбазида с концентрацией 0,5 мас.%, перемешивают и через 5-10 мин фотометрируют с зеленым светофильтром. (ПНДФ 14.1:2.52-96, Методика выполнения измерений массовой концентраций хрома в природных и сточных водах фотометрическим методом с дифенилкарбазидом, М. 1996.) Недостатком способа является его неприемлемость для анализа проб речной воды, содержащей органические и неорганические загрязнители нефтехимических производств (ориентировочный уровень концентраций загрязнителей в исходной анализируемой воде: ХПК до 50 мгО/дм3, pH от 6,0 до 8,5; остальные загрязнители в мг/дм3: взвешенные вещества до 8,0; нефтепродукты до 0,07; сухой остаток до 1000; фенолы до 0,01; аммонийный азот до 1,0; азот нитратный до 1,0; азот нитритный до 0,1; фосфор до 0,2; сульфаты до 300; хлориды до 100; цинк до 0,015; алюминий до 0,1; железо до 0,5; формальдегид до 0,1), анализируемых предварительным концентрированием и минерализацией, в результате чего происходит концентрирование содержащихся примесей до уровня, мешающего данным способом. Сущностью изобретения является обработка сконцентрированной и минерализованной пробы персульфатом аммония и серной кислотой, взятыми в количествах из расчета (0,015-0,02) г и (0,0005 - 0,0006) г моль, соответственно, независимо от объема пробы), осаждением металлов при pH=8 и последующим фильтрованием (в отличие от прототипа, где фильтрование проводят при кислотности, соответствующей 0,0006 г
моль), (5-10) см3 0,1% раствора персульфата аммония (от 0,005 до 0,01 г), кипятят 20-25 мин, фильтруют, упаривают фильтрат до объема 50 см3, нейтрализуют до pH=8, приливают 1 см3 серной кислоты (1:1) и 0,3 см3 концентрированной фосфорной кислоты, доливают раствор до 100 см3 дистиллированной водой, перемешивают, добавляют 2 см3 раствора дифенилкарбазида с концентрацией 0,5 мас.%, перемешивают и через 5-10 мин фотометрируют с зеленым светофильтром. (ПНДФ 14.1:2.52-96, Методика выполнения измерений массовой концентраций хрома в природных и сточных водах фотометрическим методом с дифенилкарбазидом, М. 1996.) Недостатком способа является его неприемлемость для анализа проб речной воды, содержащей органические и неорганические загрязнители нефтехимических производств (ориентировочный уровень концентраций загрязнителей в исходной анализируемой воде: ХПК до 50 мгО/дм3, pH от 6,0 до 8,5; остальные загрязнители в мг/дм3: взвешенные вещества до 8,0; нефтепродукты до 0,07; сухой остаток до 1000; фенолы до 0,01; аммонийный азот до 1,0; азот нитратный до 1,0; азот нитритный до 0,1; фосфор до 0,2; сульфаты до 300; хлориды до 100; цинк до 0,015; алюминий до 0,1; железо до 0,5; формальдегид до 0,1), анализируемых предварительным концентрированием и минерализацией, в результате чего происходит концентрирование содержащихся примесей до уровня, мешающего данным способом. Сущностью изобретения является обработка сконцентрированной и минерализованной пробы персульфатом аммония и серной кислотой, взятыми в количествах из расчета (0,015-0,02) г и (0,0005 - 0,0006) г моль, соответственно, независимо от объема пробы), осаждением металлов при pH=8 и последующим фильтрованием (в отличие от прототипа, где фильтрование проводят при кислотности, соответствующей 0,0006 г 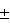
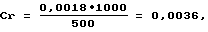 где 500 - объем пробы воды, взятый на анализ, см3; 1000 - коэффициент пересчета в дм3. Пример 3. Анализируют искусственную смесь в соответствии с изобретением (к 1 дм3 воды реки Тунгуча, содержащей 0,005 мг/дм3 хрома по результатам анализа в соответствии с примером 1, добавлено 0,003 мг хрома). Ожидаемая концентрация хрома 0,008 мг/дм3. Анализ проводят в соответствии с примером 2, отбирая на анализ 250 см3 пробы. Результат анализа: 0,0075 мг/дм3,
где 500 - объем пробы воды, взятый на анализ, см3; 1000 - коэффициент пересчета в дм3. Пример 3. Анализируют искусственную смесь в соответствии с изобретением (к 1 дм3 воды реки Тунгуча, содержащей 0,005 мг/дм3 хрома по результатам анализа в соответствии с примером 1, добавлено 0,003 мг хрома). Ожидаемая концентрация хрома 0,008 мг/дм3. Анализ проводят в соответствии с примером 2, отбирая на анализ 250 см3 пробы. Результат анализа: 0,0075 мг/дм3,Формула изобретения
Способ определения микроколичеств общего хрома в воде, включающий выпаривание пробы и последующую минерализацию, нейтрализацию и обработку минерализованной пробы персульфатом аммония в присутствии серной кислоты, фильтрование, обработку фильтрата дифенилкарбазидом в присутствии серной и фосфорной кислот и фотометрирование окрашенного раствора, отличающийся тем, что обработку персульфатом аммония в присутствии серной кислоты проводят при их количествах, взятых из расчета 0,015-0,02 г 0,0005-0,0006 гмоль соответственно на каждые 100 см3 исследуемой пробы, взятой на выпаривание, а последующее фильтрование проводят после нейтрализации до рН 8.www.findpatent.ru
Определение общего хрома в питьевой и пресной воде хемилюминисцентным методом
Государственное санитарно-эпидемиологическое нормирование Российской Федерации
4.1. МЕТОДЫ КОНТРОЛЯ. ХИМИЧЕСКИЕ ФАКТОРЫ
Определение концентрации общего хрома в питьевой и пресной воде хемилюминисцентным методом
МУК 4.1.967-99
Минздрав России
Москва 2000
1. Методические указания разработаны Федеральным центру госсанэпиднадзора Минздрава Российской Федерации (Н. С. Ластенко, И. В. Брагина, В. Б. Скачков) и ВАХЗ, ЭНТЦ «ЭкМОС» (В. А. Ишутин, А. А. Стехин, И. А. Пушкин, Г. В. Яковлева, А. А. Симонов)
2. Утверждены и введены в действие Главным государственным санитарным врачом Российской Федерации Г. Г. Онищенко 22 марта 2000 г.
3. Введены впервые.
Область применения
Методические указания по определению концентраций химических веществ в воде предназначены для использования в работе органами государственного санитарно-эпидемиологического надзора при осуществлении государственного контроля за соблюдением требований к качеству воды централизованного хозяйственно-питьевого водоснабжения водохозяйственными организациями, производственными лабораториями предприятий, контролирующими состояние водных объектов, а также научно-исследовательскими институтами, работающими в области гигиены водных объектов.
Методические указания разработаны в соответствии с требованиями ГОСТа 8.563 «Методики выполнения измерений», ГОСТа 17.0.0.02-79 «Охрана природы. Метрологическое обеспечение контроля загрязнения атмосферы, поверхностных вод и почвы».
Методики выполнены с использованием современных хемилюминесцентных методов исследования и дают возможность контролировать содержание химических веществ на уровне и меньше их предельно допустимых концентраций в воде, установленных в СанПиН 2.1.4.559-96 «Питьевая вода. Гигиенические требования к качеству воды централизованных систем питьевого водоснабжения. Контроль качества»
УТВЕРЖДАЮ
Главный государственный
санитарный врач
Российской Федерации
Г. Г. Онищенко
МУК 4.1.967-99
22 марта 2000 г.
Дата введения: 1 июня 2000 г.
4.1. МЕТОДЫ КОНТРОЛЯ. ХИМИЧЕСКИЕ ФАКТОРЫ
Определение концентрации общего хрома в питьевой и пресной воде хемилюминисцентным методом
Методические указания по определению концентрации ионов общего хрома в питьевой и пресной природной воде хемилюминесцентным методом в диапазоне концентрацией от 0,05 до 1 мг/дм3. Чувствительность метода составляет 1·10-3 мкг.
1. Метод измерения
Методика основана на измерении интенсивности хемилюминесценции реакционной смеси с помощью прибора ЛИК в момент смешения рабочего реактива с анализируемой пробой, содержащей ионов хрома. Зависимость интенсивности хемилюминесценции реакционной смеси от концентрации ионов Сr в анализируемой пробе в указанном выше диапазоне его концентраций подчиняется линейному уравнению.
Концентрация ионов Cr в исследуемой пробе воды определяется по формуле:
С = J / k,
где k - тангенс угла наклона градуировочного графика к оси абсцисс;
J - величина сигнала анализируемой пробы, мкА.
2. Нормы погрешности измерений
Допускаемая относительная суммарная погрешность результата не должна превышать 15 %. Остальные нормативы контроля метрологических характеристик определяются по результатам метрологической аттестации МВИ и приводятся в соответствующем Свидетельстве об аттестации методики.
3. Средства измерений, реактивы, материалы и посуда
3.1. Средства измерений
|
Хемилюминометр ЛИК |
ТУ 000000 АЖП |
|
рН-метр |
ГОСТ 15150-69 |
|
Дозаторы пипеточные |
ТУ 64-1-3329 |
|
Колбы стеклянные мерные со шлифами вместимостью 50, 100, 500 см3 |
ГОСТ 1770 |
|
Колбы стеклянные конические на 50 и 100 см3 |
ГОСТ 1770 |
|
Стаканы стеклянные лабораторные на 50, 100, 150 см3 |
ГОСТ 7148 |
|
Пробирки химические 10/150 |
ГОСТ 1770 |
|
Мерный цилиндр емкостью 50 см3 |
ГОСТ 1770 |
3.2. Реактивы
|
Реактив на основе люминола |
ГО 33.10.000.01 |
|
Кислота серная (азотная) концентрированная |
ГОСТ 4204-77 |
|
Натрий серноватистокислый |
СТ СЭВ 223 |
|
Стандартный образец состава раствора ионов хрома ГСО |
|
|
Хром азотнокислый |
|
3.3. Посуда
Бутылки стеклянные или полиэтиленовые с пробками на 500 см3
Фарфоровая кружка (стакан) вместимостью 100-150см3
3.4. Материалы
Бумага фильтровальная ГОСТ 12026
4. Требования безопасности
При выполнении анализов следует соблюдать правила техники безопасности при работе в химической лаборатории, включая правила работы с электроприборами напряжения до 1000 В.
5. Требования к квалификации оператора
Анализ по данной методике может проводить специалист, прошедший соответствующую подготовку.
6. Условия выполнения измерений
Измерения проводят при температуре анализируемой пробы воды 20 + 15 °С, атмосферном давлении 730-760 мм рт. ст. в лабораторных и полевых условиях.
7. Построение градуировочного графика
7.1. Подготовка воды для разведений
Разведение соли Cr готовят на водопроводной воде. Для этого открывают водопроводный кран и через 10 мин после истечения из него воды набирают ее в чистую емкость объемом 2 дм3. Емкость закрывают фильтровальной бумагой или салфеткой и дают отстоять воде при комнатной температуре в течение 24 часов.
По истечении 24 часов с помощью рН-метра определяют значение водородного показателя воды и с помощью азотной кислоты доводят его значение до рН 3,0 ± 0,1.
Внимание! После установления нужного рН проводят его повторный контроль через 5-7 минут.
7.2. Приготовление исходного разведения соли
На аналитических весах взвешивают 1,0 мг соли Cr и растворяют ее в 0,5 см3 воды, приготовленной по п. 7.1. Для этого в чистую колбу со шлифом переносят навеску взвешенной соли, наливают в нее 0,4 дм3 ± 10 см3 воды и содержимое тщательно перемешивают до полного растворения соли встряхиванием. Дают содержимому в колбе отстояться в течение 20 минут, затем колбу встряхивают в течение 3 минут, добавляя воду до метки. Исходная концентрация Cr в разведении будет равна 2,0 мг/дм3.
7.3. Приготовление разведений соли Cr для построения градуировочного графика.
Нумеруют чистые химические пробирки и устанавливают их в штатив. Дозатором пипеточным от исходного разведения отбирают 5 см3 растворенной соли в воде, переносят их в пробирку под № 1 и добавляют 5 см3, приготовленной по п. 7.1.
Содержимое закрывают пробкой и встряхивают в течение 3 минут. Полученная концентрация соли Cr составляет 1,0 мг/дм3.
Из пробирки № 1 дозатором пипеточным отбирают 5 см3 раствора соли, помещают в пробирку № 2 и добавляют туда 5 см3 воды, приготовленной по п. 7.1. Содержимое пробирки закрывают пробкой и встряхивают в течение 3 минут. Полученная концентрация соли Cr составляет 0,5 мг/дм3.
Аналогично проводят разведение соли с концентрацией 0,0625 мг/дм3.
7.4. Анализ разведенной соли Cr на приборе ЛИК
7.4.1. Готовят прибор к работе согласно инструкции.
7.4.2. Готовят рабочий реактив. Для этого в чистую пробирку наливают 10 см3 реактива и добавляют к нему 0,02 см3 3 %-ной перекиси водорода и содержимое встряхивают три раза. Реактив годен для анализа в течение 24 часов.
7.4.3. Определение фона воды, приготовленной по п. 7.1.
Крышку прибора перемещают до упора вперед, снимают с реакционной камеры крышку-дозатор. В чистую кювету наливают дозатором пипеточным 0,1 см3 рабочего реактива, помещают ее в реакционную камеру прибора и устанавливают крышку-дозатор в исходное положение. Меняют наконечник дозатора пипеточного и в полость крышки-дозатора наливают 0,2 см3 анализируемой воды.
Крышку прибора переводят до упора назад, нажимают на нее рукой и с индикатора прибора снимают его показатели. Определение проводят 3 раза, вычисляют среднее значение и результат заносят в журнал.
Внимание! При анализе разведении во избежание загрязнения реактива одна насадка дозатора пипеточного используется только для отбора реактива, а вторая - для отбора проб.
7.4.4. Анализ разведение и построение калибровочного графика.
Анализ начинают с разведения, содержащего наименьшую концентрацию искомого ингредиента и проводят его как указано в п.7.4.2. Из полученных средних значений анализа разведении вычитают фон воды и строят градуировочный график в координатах: величина измеряемого сигнала - концентрация определяемого ингредиента. График в последующем используется для определения искомого катиона в воде и уточняется (корректируется) после каждой поверки прибора.
8. Подготовка к выполнению измерений
8.1. Подготовка пробы воды к анализу
8.1.1. Пресная природная вода, вода из водоисточника. Чистый стакан емкостью 50 см3, трижды ополаскивают анализируемой водой и отбирают в него пробу объемом 40 ± 5 см3.
С помощью рН-метра определяют ее водородный показатель и устанавливают его значение в следующих пределах:
водородный показатель 3,0 ± 0,2 путем добавления серной (азотной) кислоты.
8.1.2. Питьевая вода из магистрали (крана).
Чистый стакан емкостью 500 см3 трижды ополаскивают питьевой водой из магистрали (крана), вносят в него 10 г серноватисто-кислого натрия и отбирают в него пробу объемом 500 см3.
С помощью рН-метра измеряют водородный показатель отобранной для анализа воды и устанавливают его значение в пределах, как указано в п. 8.1.1.
9. Анализ проб воды
9.1. Приготовление рабочего реактива
В чистую пробирку наливают 10,0 см3 реактива на основе люминола и добавляют к нему 0,02 см3 3 %-ной перекиси водорода. Пробирки закрывают пробкой и содержимое тщательно встряхивают в течение 2-3 минут. Срок годности рабочего реактива 24 часа с момента его приготовления.
9.2. Выполнение измерений
Готовят прибор ЛИК к работе согласно инструкции.
Крышку прибора передвигают рукой до упора вперед, снимают крышку-дозатор, наливают в кювету рабочий реактив объемом 0,1 см3, помещают ее в реакционную камеру и устанавливают крышку-дозатор в исходное положение. В полость крышки-дозатора наливают 0,2 см3 анализируемой пробы, приготовленной по п. 8.1.1 или 8.1.2, крышку рукой передвигают до упора назад, нажимают на нее и снимают показания прибора.
Анализ повторяют 3-5 раз, вычисляют среднее значение, а затем по градуировочному графику определяют искомую концентрацию ионов общего хрома в воде или рассчитывают ее из формулы
C = J/k.
10. Оформление результатов измерений
Результаты измерений оформляются протоколом по форме:
Протокол №
Протокол определения остаточного хлора
1. Дата проведения анализа ______
2. Место отбора пробы ____________
3. Название лаборатории __________
4. Юридический адрес _____________
Результаты химического анализа
|
№ пробы |
Определяемый ингредиент |
Концентрация, мг/дм3 |
Погрешность измерения, % |
|
|
|
|
|
Ответственный исполнитель
Заведующий лабораторией
11. Контроль погрешности измерения
Контроль погрешности измерения содержания в воде (хлора, железа, хрома) проводят с помощью государственного стандартного образца.
Рассчитывают среднее значение результатов измерений содержания по формуле:
 =
=  , где
, где
n - число измерений;
Сi - результат измерений;
i - число измерений в серии;
 - среднее арифметическое значение
измерений
- среднее арифметическое значение
измерений
Полученное значение должно удовлетворять условию
C - Δ  C + Δ, где
C + Δ, где
Δ - граница погрешности результата измерения, мг/дм3.
Рассчитывают среднеквадратическое отклонение результата измерений концентраций железа, введенного в воду, и выражают в единицах концентрации
S = 
и относительную квадратичную погрешность результата измерения содержания железа
S =
=
 , %
, %
Сравнивают
полученные значения отклонения результата измерений с предельно допустимыми
погрешностями. Если выполняется условие S
 Δ, то воспроизводимость измерения
является удовлетворительной. Если нет, то устраняют причины.
Δ, то воспроизводимость измерения
является удовлетворительной. Если нет, то устраняют причины.
aquagroup.ru
Опыт 7. Определение хрома
Хром образует два ряда устойчивых солей: соли хрома (III) и соли хрома (VI). Соли хрома (III) в основном содержат его в виде катиона , а соли хрома (VI) - в виде анионови. Растворы солей хрома (III) имеют сине-зеленую или фиолетовую окраску, хроматов () - желтую, а бихроматов () - оранжевую.
а) Определение катиона Сr3+ щелочью.
Катион хрома (III) образует с сильными гидроксидами (щелочами) осадок серо-зеленого цвета. При действии на осадок избытка щелочи хром переходит в раствор в виде хромит-иона зеленого цвета (цвета травы)
;
.
К 2 каплям раствора соли хрома (III) добавьте 1 каплю 2 н раствора гидроксида натрия Наблюдайте образование серо-зеленого осадка гидроксида хрома. Затем к осадку добавьте избыток щелочи до его полного растворения. Отметьте цвет осадка и раствора. Напишите уравнения соответствующих реакций.
б) Окисление хрома (III) до хрома (VI).
Катион хрома (III) легко окисляется пероксидом водорода в щелочной среде до :
.
К 2 - 3 каплям раствора соли хрома (III) добавьте несколько капель 2 н раствора гидроксида натрия до полного растворения образующегося вначале осадка гидроксида хрома, 2 - 3 капли 3%-ного раствора пероксида водорода и нагрейте несколько минут до изменения зеленой окраски раствора в желтую.
Проведите соответствующие реакции, отметьте исходный, промежуточный и конечный цвет раствора, напишите уравнения реакций.
в) Смещение равновесия между хромат- и бихромат-ионами.
Хром (VI) может присутствовать в растворе в виде хромат-ионов желтого или бихромат-ионоворанжевого цвета. Между ними существует равновесие
,
которое в кислой среде смещается в сторону иона , а в щелочной- иона.
К 2 - 3 каплям раствора, содержащего анион , добавьте 2 - 3 капли 2 н раствора серной кислоты. Наблюдайте изменение желтой окраски в оранжевую:
.
К 2 - 3 каплям раствора, содержащего анион , добавьте 2 - 3 капли 2 н раствора гидроксида натрия. Наблюдайте изменение оранжевой окраски в желтую:
.
г) Определение хромат- и бихромат-ионов хлоридом бария .
Хлорид бария образует с хромат- и бихромат-ионами желтый осадок хромата бария (произведение растворимости хромата бария достигается раньше, чем бихромата)
;
.
Хромат бария растворим в сильных кислотах, поэтому анализ ведут в присутствии ацетата натрия , который нейтрализует кислоту, выделяющуюся в результате реакции.
К анализируемому раствору добавьте несколько капель раствора ацетата натрия и 2 - 4 капли хлорида бария. Наблюдайте образование желтого осадка хромата бария.
Медь
Медь присутствует в воде в виде катиона . Степень ее токсичности определяется между низкой и средней.
Накопление избытка меди в организме ведет к болезни Вильсона, нарушению функций печени.
Источниками поступления меди в воду могут служить рудные воды, сточные воды гальванического производства, продукты коррозии медьсодержащих сплавов и др.
Предельно допустимая концентрация некоторых солей и органических производных меди составляет 1 мг/л.
Опыт 8. Определение меди
Растворы солей меди (II) окрашены в голубой цвет.
а) Определение щелочами.
Сильные основания образуют с катионом меди голубой осадок гидроксида меди :
.
Гидроксид меди обладает слабовыраженными амфотерными свойствами и поэтому частично растворяется, особенно при нагревании, в избытке щелочи с образованием купритов.
К 2 - 3 каплям раствора, содержащего медь (II), добавьте 2 капли 2 н раствора Наблюдайте образование голубого осадка.
б) Определение гидроксидом аммония.
Гидроксид аммония добавленный в небольшом количестве в раствор, содержащий катион меди (II), осаждает основную соль зеленоватого цвета, легко растворимую в избытке реагента. При этом образуется аммиачный комплекс меди интенсивно-синего цвета. Например,
;
.
К 2 - 3 каплям раствора, содержащего соли меди (II), осторожно добавьте по каплям гидроксид аммония до образования осадка. Затем, добавив избыток реагента, растворите этот осадок. Отметьте цвет образовавшегося осадка и раствора. Напишите уравнения соответствующих реакций.
в) Определение гексацианоферратом (II) калия .
При добавлении к раствору, содержащему соли меди (II), раствора образуется красно-бурый осадок
.
Осадок не растворяется в разбавленных кислотах, но растворяется в гидроксиде аммония и в растворах щелочей.
В раствор, содержащий соли меди, добавьте несколько капель уксусной кислоты и раствора гексацианоферрата (II) калия до появления характерного осадка. Отметьте цвет осадка. Напишите уравнение химической реакции.
studfiles.net
Хром и методы его определения
Казанский Государственный Технический Университет им. А.Н. Туполева Курсовая работа на тему: «ХРОМ И МЕТОДЫ ЕГО ОПРЕДЕЛЕНИЯ» Выполнил: ст. гр.3572 Боброва М.Г. Научный руководитель: Буданов А.Р. Проверил: Шамкаева А.И. Казань 2007Содержание Введение 1. Литературный обзор. 1.1. Современное состояние технологической линии 1.2. Характеристика твердых отходов процесса хромирования 1.3. Хром, его физические и химические свойства, хромирование 2. Методы определения 2.1. Титрование сульфатом железа (II) и перманганатом 2.2. Колометрические методы. Хроматный метод 2.3. Метод с дефинилкарбазидом 2.4. Атомно-абсорбционный метод 2.5. Другие методы 3. Теория определения хрома экспериментально. Качественный анализ компонентов твердых отходов процесса хромирования 4. Получение результатов Выводы Список литературы Приложения
Введение Хромирование начали применять в промышленности с конца двадцатых годов нашего столетия. Этот процесс существенно отличается от большинства других катодных гальванических процессов в силу ряда химических, электрических и технологических особенностей. Несмотря на некоторую свою «необычность», хромирование получило очень широкое распространение. Вряд ли можно назвать другое гальваническое покрытие, обладающее более обширным ассортиментом технически полезных свойств и используемое в более разнообразных областях промышленности. Нельзя забывать так же о том, что шламы гальванических процессов являются основным источником загрязнения почвы, водоемов и сельскохозяйственных угодий. При неэффективной очистке гальваностоков тяжелые металлы попадают в природные водоемы, почву и по пищевым цепям доходят до человека. Аналогичная ситуация возникает при выщелачивании тяжелых металлов кислотными дождями и природными органическими кислотами из шламов в местах их захоронений. Таким образом, круг замыкается, и растворы солей тяжелых металлов в конечном итоге попадают в водоемы. В настоящее время гальванические производства имеют практически все предприятия машиностроительной, электротехнической и других отраслей промышленности России. Переработка гальванических шламов для предприятий обременительна, поэтому после нейтрализации (перевода в менее растворимые соединения) направляются на захоронение. Однако это не решает проблемы сохранения окружающей среды. Поскольку и после нейтрализации шламы являются в той или иной степени токсичными. Попадание ионов тяжелых металлов в почву и воду вызывает антропогенные геохимические аномалии в атмосфере, гидросфере, приводит к ослаблению жизнедеятельности почвенных бактерий, определяющих плодородие почвы, оказывает вредное воздействие на живые организмы растительного и животного мира. В частности хром является весьма канцерогенным веществом, вызывающий опухолевые процессы. В последнее время установлено, что ионы тяжелых металлов нарушают работу кальмодумина – основного регулятора процессов жизнедеятельности организма, в результате чего развиваются наследственные болезни, сердечно-сосудистые расстройства, рак и др. К концу ХХ века из ежегодно образовывающихся в России более чем 20 млн. тонн неутилизируемых высокотоксичных промышленных отходов 0,75 млн. тонн составили гальваношламы. Несмотря на значительное снижение объемов гальванического производства в последние годы, которое по некоторым оценкам достигло 40-50 %, проблема утилизации гальванических шламов и сточных вод гальванического производства остается для Российской Федерации одной из наиболее важных. Задача: определить качественный состав компонентов твердожидких отходов гальванического производства процесса хромирования.
1. Литературный обзор 1.1 Современное состояние технологической линии хромирования Современные сложные технологические производства во всех отраслях промышленности широко используют различные гальванические процессы. Находят применение такие процессы, как нанесение покрытий, травление, электрохимические методы размерной обработки, обезжиривание и др. Реализация гальванических процессов осуществляется в ваннах, или в среде проточного электролита. В процессе эксплуатации рабочие жидкости интенсивно загрязняются продуктами электролиза в виде шлама, ионами тяжелых металлов, жировыми загрязнениями после промывочных операций. Электрохимический процесс хромирования происходит в несколько стадий. Первоначально стальную деталь химически обезжиривают путем опускания ее в ванну, содержащую смесь растворов определенного количества (см. Приложение). В качестве обезжиривателя выступает вода питьевая. В конце процесса на дне ванны остаются отходы (см. Приложение). Затем производят промывку в горячей проточной воде, после чего производят обезжиривание электрохимическое, также сопровождающееся выделением отходов. По окончании обезжиривания промывают в горячей, затем в холодной проточной воде. Далее - химическая активация, промывка в холодной проточной воде. Затем непосредственно производят электрохимическое хромирование, после чего на дне ванны остаются отходы. Процесс хромирования завершается промывкой в проточной холодной воде, после чего промывают стальную деталь в проточной горячей воде. Далее - производят транспортировку и выгрузку готовых деталей. Следует отметить, что на предприятии используются устаревшие технологические процессы, и отсутствует сортировка твердо-жидких отходов гальванических производств. Твердо-жидкие отходы, для упрощения далее называемые «твердые отходы гальванического производства», образуются при периодической зачистке ванн каждой гальванической линии (виды ванн и периодичность зачистки приведены в Приложении). При определении количества образования данного вида отходов отсутствует методологический подход к оценке их образования, полный учет их образования и точный анализ состава. 1.2 Характеристика твердых отходов процесса хромирования В работе анализировались методы определения твердо-жидких отходов гальванических производств с целью оценки качественного состава твердых отходов техпроцесса хромирование. Характеристика технологического процесса приведена в Приложении. Отходы хранятся на территории цехов в закрытых металлических емкостях по 50 кг каждая. По статистическим данным на КАПО им. С.П. Горбунова за год образуется в среднем 10 т твердых гальванических отходов в год. (Данные не уточняются ввиду того, что отходы с основных ванн технологических процессов не сортируются). 1.3 Хром, его физические и химические свойства, хромирование Хром – серебристый с голубоватым отливом металл. Атомный вес его 52, удельный вес 7,1 г/см3, температура плавления 1830 оС, электрохимический эквивалент 0,323 г/а-ч, нормальный потенциал 0,55 в. Благодаря склонности к пассивации хром длительное время сохраняет блеск. Он стоек во влажной атмосфере, к действию щелочей, органических кислот, серы, серной и азотной кислот. В гальванической паре с железом хром является катодом, несмотря на более электроотрицательный потенциал. Пассивная пленка, покрывающая хром, сдвигает его потенциал в положительную сторону, а значительная пористость хромовых покрытий вызывает необходимость создания подслоя для защиты стальных изделий. Хром обладает рядом ценных качеств. Кроме упомянутой уже химической стойкости и способности длительное время сохранять блеск, хрому свойственна высокая твердость, низкий коэффициент трения, значительная жаростойкость. Эти свойства хромовых покрытий делают гальваническое хромирование весьма распространенным видом покрытия, которое практически нельзя заменить другим. Основные области применения хрома – декоративная защита, повышение износостойкости трущихся деталей, поршневых колец и цилиндров, мерительного и режущего инструмента, коррозионно-стойкие комбинированные покрытия. Декоративные хромовые покрытия осаждают на предварительно никелированные или омедненные и никелированные покрытия. Разработаны электролиты, позволяющие непосредственно на стали получать малопористые хромовые покрытия, обладающие защитными и декоративными свойствами. Технология получения комбинированного двухслойного хрома позволяет сочетать в покрытии коррозионно-стойкие и защитные функции. Хромовые осадки отличаются высокими внутренними напряжениями, поэтому на хромовых покрытиях уже при толщине слоя 0,1 мк образуется сетка мелких трещин. При увеличении толщины хромового покрытия старые трещины перекрываются вновь образовавшимися. В зависимости от условий хромирования сеть трещин может быть различной. Это свойство хромовых покрытий используют для предания деталям антифрикционных свойств, а также для создания новой технологии получения комбинированного защитно-декоративного никель-хромового покрытия. Хромовые покрытия в 5-8 раз повышают износостойкость стали. Самой высокой износостойкостью отличаются матовые хромовые покрытия, самой низкой – блестящие. Наибольшее влияние на твердость хромовых покрытий имеет режим электролиза – температура электролита и применяемая плотность тока.
2. Методы определения Наиболее удовлетворительными методами определения хрома являются колориметрические и объемный. Объемный метод основан на окисление хрома до хромата, прибавление избыточного количества сульфата железа (II) и титрование избытка последнего перманганатом. Колориметрический метод пригоден для определения малых количеств хрома, какие обычно содержаться в горных породах. При значительном содержании хрома, когда колориметрические методы не применимы, пользуются объемным методом. 2.1 Титрование сульфатом железа(II) и перманганатом По нашим данным, наиболее удовлетворительный метод определения хрома заключается в окислении хрома до хромата, введение в раствор отмеренного количества сульфата железа (II) и оттитровывании избытка последнего перманганатом или бихроматом калия. Окисление хрома можно осуществлять в процессе сплавления или соответствующей обработкой кислого раствора. Оба эти метода нашли практическое применение. Сплавлением с карбонатом натрия и нитратом калия пользуются при определении малых количеств хрома; когда определяют большие количества хрома, лучше применять сплавление с перекисью натрия. В кислом растворе хром можно окислить двуокисью свинца, хлоратом калия, перманганатом калия и персульфатом калия или аммония в присутствии нитрата серебра. Из этих методов наилучшим является последний. Ниже приводится его подробное описание. В персульфатном методе присутствие нитрата серебра имеет существенное значение для полного окисления хрома. Нитрат серебра вводят в горячий кислый раствор перед прибавлением персульфата так, чтобы его содержание было не меньше, чем содержание хрома. При чрезмерно высокой концентрации кислоты в растворе достигнуть полного окисления хрома не удается. Наиболее благоприятными условиями является содержание 5-6 мл серной и 1 мл азотной кислот в 100 мл раствора. В присутствии железа и вольфрама целесообразно вводить 3-5 мл сиропообразной фосфорной кислоты, так как она удерживает вольфрам в растворе и, кроме того, обесцвечивая желтую окраску железа (III), делает конечную точку титрования более отчетливой. Для окисления применяют свежеприготовленный раствор персульфата 95%-ной чистоты. Во всех случаях анализируемый раствор следует кипятить 8-10 минут, чтобы обеспечить полное окисление хрома и разложение избытка персульфата. Марганец при этом окисляется до перманганата или двуокиси марганца, которые также необходимо разрушить до прибавления сульфата железа (II). Для этого, после разложения персульфата кипячением, на каждые 300 мл раствора прибавляют по 5 мл разбавленной (1:3) соляной кислоты и продолжают кипятить до восстановления окисленных соединений марганца, после чего кипятят еще 5 минут для удаления хлора. Если до прибавления соляной кислоты персульфат был полностью разрушен, хромовая кислота при этой обработке не восстанавливается. Большие количества хрома наиболее целесообразно восстанавливать взвешанным количеством однородных кристаллов соли Мора FeSO4(Nh5)2SO4*6h3O. В случае же, когда содержание хрома невелико и особенно когда приходится одновременно проводить большое число определений, удобнее пользоваться разбавленным сернокислым раствором этой соли. В присутствии значительных количеств хрома определение конца титрования несколько затрудняется вследствие зеленой окраски раствора, но соответствующая поправка для конечной точки определяется легко. Все реакции протекают стехиометрически; следовательно, можно пользоваться теоретическими титрами, установленными по оксалату натрия. Ванадий и мышьяк не мешают титрованию, так как они хотя и восстанавливаются сульфатом железа (II), но затем снова окисляются эквивалентным количеством перманганата. Небольшие количества вольфрама изменяют окраску раствора в точке эквивалентности, но не влияют на результаты титрования. Если содержание вольфрама настолько велико, что конечная точка титрования становится неотчетливой, его лучше предварительно отделить, а хром, захваченный осадком вольфрама, определить отдельно. Никель, кобальт, молибден и уран не оказывают влияния на титрование. Ход определения. Раствор, полученный после разложения пробы с плавлением или другим способом, свободный от хлорид-ионов и содержащий приблизительно 15-18 мл серной кислоты и 3 мл азотной кислоты в общем объеме 300 мл, нагревают до кипения. Прибавляют 2,5%-ный раствор нитрата серебра в количестве, соответствующем 0,01 г соли на каждую 0,01 г находящегося в растворе хрома. Нагревают до кипения и приливают 20 мл свежеприготовленного 10%-ного раствора персульфата аммония. Кипятят 10 минут и затем, если образуется перманганат или окислы марганца, вводят 5 мл 5%-ного раствора хлорида натрия или 5 мл разбавленной (1:3) соляной кислоты, снова нагревают до кипения и после восстановления соединении марганца продолжают кипятить еще 5 минут. Если при этом марганец не восстанавливается, вводят еще некоторое количество хлорида натрия или соляной кислоты и снова кипятят. После охлаждения раствора прибавляют отмеренное избыточное количество раствора соли Мора или взвешенное количество этой соли и затем титруют раствором перманганата, титр которого установлен по оксалату натрия. По окончании титрования отмечают израсходованное количество перманганата, нагревают раствор до кипения для разрушения избытка перманганата, охлаждают до комнатной температуры и затем снова титруют перманганатом до появления окраски такой же интенсивности, как при первом титровании. Израсходованный в этом случае объем перманганата вычитают из объема раствора, израсходованного при первом титровании. Полученную разность умножают на количество соли Мора, эквивалентное 1мл раствора перманганата. Это произведение вычитают из введенного количества соли Мора и затем вычисляют содержание хрома, исходя из соотношения 3Fe:1Cr. Для установления соотношения между солью Мора и перманганатом приготавливают раствор из такого же количества соли, какое было введено в анализируемый раствор; приготовленный раствор должен иметь такой же объем и такую же кислотность, как анализируемый раствор в момент титрования. Приготовленный раствор титруют перманганатом. Из израсходованного объема раствора перманганата вычитают количество его, которое требуется для окрашивания раствора в точке эквивалентности. При выполнении массовых анализов после сплавления с перекисью натрия нет необходимости окислять хром персульфатом. В этом случае плав растворяют в 200 мл воды, прибавляют 1г перекиси натрия и сильно кипятят до разложения перекиси (приблизительно 10 минут). Фильтруют через асбест и промывают осадок горячей водой, следя за тем, чтобы он все время был влажным. Фильтрат и промывные воды подкисляют серной кислотой и затем вводят сульфат железа (II). При этом следует избегать введения в раствор органических веществ, (что происходит, например, при фильтровании через бумагу), так как это приводит к получению пониженных результатов для хрома. Эта ошибка обусловлена не восстановлением хромата, как можно было бы предположить, а восстановлением перманганата в процессе титрования. Легкость, с которой можно осуществить потенциометрическое титрование хрома, применяя соответствующую аппаратуру, характеризуется следующим ходом анализа. В охлажденный до 20С разбавленный сернокислый раствор, содержащий хром (+6), погружают каломельный и платиновый электроды; пучок лучей, отраженных зеркальным гальванометром, устанавливают вблизи левого края шкалы. Затем в раствор постепенно вводят титрованный раствор сульфата железа (II), пока пучок лучей не начнет непрерывно передвигаться вправо. После этого прибавляют эквивалентный титрованный раствор хромата калия до прекращения перемещения пучка лучей влево. Снова вводят раствор сульфата железа (II), пока опять не начнется перемещение вправо. Вычитают израсходованный объем раствора хромата из введенного объема сульфата железа и вычисляют содержание хрома, исходя из соотношения 1Cr:3Fe. Ванадий также восстанавливается железом (II) и учитывается совместно с хромом. Вольфрам не влияет на титрование.
2.2 Колориметрические методы. Хроматный метод Определение хрома колометрическим методом преимущественно проводят сравнением интенсивности окраски хромата с окраской стандартного раствора в щелочной среде. При анализе горных пород для этого обычно используют водную вытяжку плава анализируемого материала со смесью карбоната натрия и нитрата калия. Обычно встречающиеся в горных породах элементы определению не мешают. При плавлении пробы в платиновом тигле не следует вводить в плавень слишком больших количеств селитры и температура сплавления не должна быть слишком высокой, чтобы не вызвать порчу тигля, так как раствор может окраситься в желтый цвет за счет переходящей в него платины. При сплавлении с перекисью натрия и выщелачивании плава водой некоторое количество железа может остаться в растворе и окрасить его. Этого можно избежать, если перед фильтрованием нагревать раствор на водяной бане не менее одного часа. Щелочной раствор окрашивается также в случае фильтрования его через бумажный фильтр, что, однако, может быть устранено, если предварительно тщательно промывать применяемый фильтр раствором щелочи или, лучше, применением для фильтрования этого раствора асбеста. Бораты не мешают определению. Уран в условиях колориметрирования хрома также дает желтую окраску, и, следовательно, в его присутствии метод не применим. Наиболее удобно сравнивать окраски растворов, содержащих приблизительно 0,01 – 0,1 мг Cr2O3 в одном мл. Ход определения. Приготовляют стандартный раствор бихромата, содержащий 0,1 мг Cr2O72- в 1мл. Для этого 0,1934 г перекристаллизованного бихромата калия K2Cr2O7 растворяют в воде и разбавляют до 1 л. Перед применением этого раствора для сравнения с анализируемым его следует сделать щелочным. Для определения хрома в горных породах 1 –5 г пробы сплавляют со смесью карбоната и селитры. Плав растворяют в воде, и если в растворе находится перманганат, прибавляют несколько капель этилового спирта для разрушения его окраски, а затем фильтруют через асбест. Если окраска раствора чрезмерно бледна, повышают концентрацию хрома упариванием раствора или осаждением нитратом ртути (I) и последующей обработкой. При анализе других объектов, например осадков от аммиака, содержащих малые количества хрома, окисление можно осуществлять сплавлением с перекисью натрия в фарфоровом тигле. Во всех случаях полученный раствор хромата переносят в мерную колбу такой емкости, чтобы интенсивность окраски раствора после разбавления до метки соответствовала 0,01-0,1 мг Cr2O3 в 1 мл. Заканчивают определение титрованием раствора такого же количества реактивов стандартным раствором бихромата калия до одинаковой с анализируемым раствором окраски или же каким-нибудь другим обычно применяемым колориметрическим или фотометрическим способом измерения. 2.3 Метод с дефинилкарбазидом Малые количества хрома (микрограммы) можно определять по реакции с дефинилкарбазидом, который окисляется бихроматом в слабокислом растворе с образованием соединения, окрашенного в красно-фиолетовый цвет. Ванадий, если он присутствует в значительных количествах, мешает определению. Его можно удалить экстракцией хлороформом после переведения в оксихинолят. Уран не влияет на реакцию с дефинилкарбазидом. 2.4 Атомно-абсорбционный метод Сущность метода. Прямое определение хрома возможно, когда его концентрация превышает 100 мкг/л. Если приходиться анализировать более разбавленные растворы, то во многих случаях достаточно упарить раствор после подкисления его азотной кислотой; но при анализе очень разбавленных растворов или при необходимости повысить чувствительность определения рекомендуется предварительно выделить металл экстракцией. Реактивы. Горючие газы – ацетилен, пропан, водород. Можно пользоваться продажными баллонами, снабженными редукторами. Воздух. Должен быть отделен от посторонних веществ пропусканием через фильтр и высушиванием. Деионизированная дистиллированная вода. Ее следует применять при приготовлении всех реактивов, калибровочных стандартных растворов и при разбавлении пробы. Соляная кислота, концентрированная. Азотная кислота, концентрированная. Стандартные растворы металлов. Приготавливают серии стандартных растворов солей различных металлов, концентрацией 5 – 1000 мкг/л, соответствующим разбавлением запасных растворов дистиллированной водой, содержащей 1,5 мл концентрированной азотной кислоты в одном литре. Запасные растворы солей. Хром. Растворяют 2,8289 г K2Cr2O7 в 200 мл дистиллированной воды, прибавляют 1,5 мл концентрированной HNO3 и разбавляют до 1000 мл такой же водой; 1,00 мл полученного раствора содержит 1 мл хрома. Ход анализа. Приборы для атомно-абсорбционной спектрофотометрии различают и по конструкции, и по методике работы на них, поэтому следует строго следовать прилагаемой к прибору инструкции. Приводим лишь некоторые ступени хода анализа. Вставляют пустотелую катодную лампу, предназначенную для определения требуемого элемента, и устанавливают на указанную для определения этого элемента длину волны (хром: длина волны – 357,9 нм; горючий газ – ацетилен; газ-окислитель – воздух). Определяют оптимальное соотношение горючего газа и газа-окислителя, измеряя отношение в области, близкой к ориентировочным данным, и отмечают отношение с минимальным поглощением при холостом опыте и с максимальным поглощением определяемого элемента – хрома. Концентрацию последнего выбирают так, чтобы абсорбция была 0,5-0,8. Определяют время достижения равновесного состояния с момента впрыскивания пробы. Находят оптимальную ширину щели, определяют оптимальную высоту оптической оси над горелкой, выявляя максимум абсорбции стандартного раствора при перемещении горелки в вертикальном направлении. Для построения градуировачного графика вводят поочередно в пламя горелки рабочие стандартные растворы, начиная от раствора с минимальным содержанием определяемого элемента: не менее четырех концентраций, включая концентрацию, близкую к той, которая ожидается в анализируемом растворе. Каждое измерение проводят не менее двух раз, при построении графика берут среднее значение. 2.5 Другие методы Хром количественно осаждается аммиаком. Осадок следует под конец прокаливать в атмосфере водорода, иначе получаются повышенные результаты вследствие окисления хрома в процессе прокаливания. В связи с этим, а также и потому, что хром почти всегда сопровождают посторонние, осаждающиеся аммиаком элементы, как, например, железо, алюминий, фосфор и ванадий, этим методом для определения хрома пользуются лишь в редких случаях. Осаждение хрома в виде хромата серебра Ag2CrO4, хромата ртути Hg2CrO4 и хромата бария BaCrO4 представляет интерес главным образом для группового разделения и качественного испытания на хром, а не для количественного его определения, так как многие другие элементы также образуют нерастворимые соединения с этими реагентами. Точные результаты получаются при определении хрома методом, основанным на восстановлении хромата иодистоводородной кислотой и титровании выделяющегося при этом йода раствором тиосульфата натрия. Этот метод, однако, не получил такого широкого распространения, так как железо, медь, мышьяк, ванадий и молибден, которые в состоянии высшей валентности выделяют йод в кислых растворах йодида калия, должны отсутствовать. Известен колориметрический метод определения хрома с комплексоном III (этилендиаминтетраацетатом натрия). Метод специфичен, мешает только окрашенные катионы (своей окраской), но сравнительно мало чувствителен (оптимальные концентрации хрома 5-80 мг/л). Светопоглощение получаемого красно-фиолетового раствора измеряют, применяя зеленые светофильтры (длина волны 550 нм).3. Теория определения хрома экспериментально. Качественный анализ компонентов процесса хромирования Объект исследования: твердые отходы гальванических процессов. Оборудование и реактивы: пробирки, фарфоровая ступка, пестик, растворы кислот, щелочей, вода дистиллированная, химические стаканы, стеклянная палочка, сухое горючее, складчатый фильтр. Поступившую для анализа пробу необходимо измельчить в фарфоровой ступке, тщательно перемешать и взять среднюю пробу. Среднюю пробу необходимо отобрать методом квадрата (пробу разложить в виде квадрата на листе белой бумаги и делить диагоналями на четыре треугольника, две противоположные части отбрасываются, а две другие соединяются, снова ссыпаются в фарфоровую ступку и еще раз измельчаются и снова делится квадрат по диагонали). Полученную таким образом среднюю пробу помещают в банку с притертой пробкой. Затем производят процессы разложения (вскрытия) пробы, растворение. растворение в воде: небольшое количество средней пробы помещают в пробирку и растворяют в дистиллированной воде при комнатной температуре. С помощью универсальной Ind бумаги определяют характер среды pH и растворение осадка в воде. проводится также растворение пробы небольшого количества при нагревании, оценивается осадок и среда рН. растворение в трех мл 2н серной кислоте 0,02 г пробы: наблюдается растворение осадка, изменение цвета окраски, pH среды. растворение в Nh5Cl, Nh5OH также наблюдаются изменения. растворение в NaOH (8% и 4н) изменение окраски осадка. 4. Получение результатов В результате эксперимента делают предположения о наличии ионов железа, алюминия и др., которые подтверждаются специфическими реакциями. Отфильтровав осадок и промыв его дистиллированной водой, растворяют в различных кислотах и щелочах, добавляют специфические реагенты и наблюдают аналитический сигнал. Если при растворении твердого отхода в HNO3 (конц), добавить персульфат аммония (Nh5)2S2O8, наблюдается окрашивание раствора в оранжевый цвет, то это говорит о наличии ионов Cr+6, т.е. реакция восстановления ионов хрома 6+ до хрома3+ не произошла или произошла не до конца.
Выводы Отходы с основных ванн гальванического производства смешиваются, экспериментальные данные могут показать заниженное содержание тяжелых металлов, учитывая это пробы необходимо брать в достаточном количестве и, если это возможно, то отход гальванического производства необходимо отобрать от конкретного технологического процесса.
Список литературы 1. М.П. Грачева «Гальванотехника при изготовлении предметов бытового назначения» 2. Лурье Ю.Ю. «Практическое руководство по неорганическому анализу» - М.,1960 г. 3. «Вредные вещества в промышленности. Справочник для химиков, инженеров, врачей» под ред. Лазарева Н.В., Гадаскиной И.Д., 608стр. 4. Крешков А.П. «Основы аналитической химии. Теоретические основы. Качественный анализ» - М.,1970 г. 5. Журнал «Вестник Татарстанского отделения Российской Экологической Академии», 1(19) 2004 г. 6. «Охрана окружающей среды от отходов гальванического производства» Ю.Н. Меркушев, В.Г. Маклецов, В.Г. Петров (материал семинара) – М.:1990 г. ПРИЛОЖЕНИЕ Состав твердожидкого отхода
| № п/п | Технологическая линия | Ванна, с которой удаляется твердо-жидкий отход | Теоретический отход | Основные параметры |
| 1. | Хромирование | Обезжиривание химическое и электрохимическое | 90%-«органика»10%-М(ОН)2 ,MeSiO3 , Me2 (PO4)3 ,Me=Fe,Cu | Объем ванны: 2500 л. Периодичность очистки – 1 раз в 2 годаКоличество ванн: 1 шт. |
| Хромирование стальных деталей | Fe(OH)3, Fe(OH)2, FeS Cr(OH)3-50% Cr+6, сульфаты K2SiF6, BaSO4, Fe, Cu- до 10г/л | Объем ванны: 2500 л. Периодичность очистки – 1 раз в 2 годаКоличество ванн: 2 рабочие ( всего 3шт.) | ||
| Активация в кислоте стальных деталей | Соединение железа – 40% | Объем ванны: 980 л.Периодичность очистки – 1 раз в 2 годаКоличество ванн: 1 шт. |
| Обезжиривание химическое и электрохимическоеNaOH 5-15 г/лNa3PO4 30-70 г/лNa2SiO3 10-20 г/лNa2CO3 20-25 г/л |
| Промывка в горячей проточной воде |
| Промывка в холодной проточной воде |
| Активация h3SO4 50-100 г/л |
| Промывка в проточной воде |
| ХромированиеCrO3 – хромовый ангидрид 250 г/лh3SO4 2.5% от количества CrO3 |
baza-referat.ru
способ определения микроколичеств общего хрома в воде - патент РФ 2137112
Изобретение относится к области аналитической химии, а именно к способу определения общего хрома (CrIII и CrVI) в воде рыбохозяйственных водоемов с нормой по содержанию хрома не более 0,001 мг/дм3. Способ включает предварительное концентрирование хрома путем выпаривания и обработку, позволяющую устранить мешающее влияние примесей фотометрическому определению хрома по реакции с дифенилкарбазидом. Изобретение относится к области аналитической химии, а именно к способу определения общего содержания хрома (CrIII и CrVI) в воде рыбохозяйственных водоемов с содержанием хрома в пределах 0,001 до 0,01 мг/дм3. Известен способ определения хрома методом пламенной атомно-абсорбционной спектрометрии (ПНДФ 14.1:2:22-95. Количественный химический анализ вод. Методика выполнения измерений массовой концентрации железа, кадмия, свинца, цинка и хрома в пробах природных и сточных вод методом пламенной атомно-абсорбционной спектрометрии), чувствительность метода 0,02 мг/дм3. Недостатком способа является его недостаточная чувствительность для анализа вод с нормой по содержанию хрома "не более 0,001 мг/дм3", к каким могут быть отнесены пробы воды с показателем pH от 7,0 до 8,5, содержащих восстановители и (или) окрашенные органические вещества, в которых из-за невозможности раздельного определения CrIII и CrVI, определяют только общее содержание хрома, однако, для санитарно-гигиенической оценки качества воды расчеты ведут по наиболее токсичному (ПДК = 0,001 мг/дм3) хрому VI. (Ю.В. Новиков и др., Методы исследования качества воды водоемов, Изд. "Медицина", М. 1990, с. 228). Известен способ фотометрического определения малых количеств суммарного содержания хрома в водах, имеющих щелочную реакцию или содержащих большие количества органических веществ, хлоридов, по которому в пробе, содержащем от 0,001 до 0,05 мг хрома, сначала восстанавливают хром (VI) до хрома (III) сернистой кислотой, затем осаждают хром (III) оксидом магния, отфильтровывают, высушивают, сжигают, растворяют в серной кислоте, окисляют хром (III) до хрома (VI) персульфатом аммония или прокаливанием со смесью карбоната натрия и оксида магния, приливают раствор дифенилкарбазида и фотометрируют. (Ю. Ю. Лурье. Аналитическая химия промышленных сточных вод, Изд. "Химия". М. 1984. с. 155.). Недостатком способа является трудоемкость и высокая погрешность определения, связанная с его многостадийностью, и недостаточная чувствительность (0,02 мг/дм3) для анализа природных вод с нормой по содержанию хрома 0,001 мг/дм3. Известен способ определения хрома в промышленных сточных водах рентгенофлуоресцентным методом (ПНДФ 14.1.43.96. Количественный химический анализ вод. Методика выполнения измерений массовой концентрации ванадия, хрома, марганца, железа, кобальта, никеля, меди, цинка, свинца и висмута в промышленных сточных водах рентгенофлуоресцентным методом), чувствительность метода 0,01 мг/дм3. Недостатком способа является его низкая чувствительность и необходимость дорогостоящего специального оборудования. Известен способ определения общего хрома в воде с помощью атомно-абсорбционной спектрометрии при электротермической атомизации пробы (Г.С. Фомин, Вода. Контроль химической, бактериальной и радиационной безопасности по международным стандартам. Энциклопедический справочник. ИСО 9174, Метод "В",), чувствительность метода 0,005 мг/дм3. Недостатком способа является его неприемлемость для аналитического контроля воды рыбохозяйственных водоемов, для которых предельно допустимая концентрация (ПДК) по содержанию хрома составляет 0,001 мг/дм3. Известен способ определения общего хрома в пробах природной питьевой и сточной воды на анализаторе Флюорат-2, основанный на регистрации свечения хемилюминесценции реакции окисления люминола раствором пероксида водорода, катализируемого солями хрома (III) в щелочной среде (ПНДФ 14.1:2:4. 30-95, Количественный химический анализ вод. Методика выполнения измерений массовой концентрации хрома общего в пробах природной, питьевой и сточной воды на анализаторе жидкости "Флюорат-02"), Минимально определяемая концентрация 0,002 мг/дм3. Недостатком способа является необходимость применения дорогостоящего специального оборудования. Известен способ определения общего хрома в пробах питьевых природных и сточных вод методом инверсионной вольтамперометрии, основанный на адсорбционном концентрировании на поверхности графитового электрода 1,5-дифенилкарбозоната хрома (III), образовавшегося в результате реакции дифенилкарбозона с ионами Cr(VI). Минимально определяемая концентрация хрома 0,001 мг/дм3 (ПНДФ 14.1:2:4.72-96, Количественный химический анализ вод. Методика выполнения измерений массовой концентрации хрома в пробах питьевых, природных и сточных вод методом инверсионной вольтамперометрии.). Недостатком способа является его неприемлемость для анализа вод, содержащих органические загрязнения (содержание органического углерода не должно превышать 0,001 мг/дм3) и необходимость применения специфического оборудования. Наиболее близким по технической сущности и достигаемому результату является способ фотометрического определения общего содержания хрома в природных и сточных водах по реакции хрома (VI) с дифенилкарбазидом, по которому отбирают такой объем исследуемой пробы (разбавленной, сконцентрированной упариванием, минерализованной), в котором содержится (0,001-0,05) мг хрома, доводят объем до 100 см3, нейтрализуют пробу до pH=8 кислотой или щелочью, добавляют 0,3 см3 раствора серной кислоты с концентрацией с(1/2h3SO4)=1 моль/дм3 (1: 2:1000 0,3=0,0006 г моль), (5-10) см3 0,1% раствора персульфата аммония (от 0,005 до 0,01 г), кипятят 20-25 мин, фильтруют, упаривают фильтрат до объема 50 см3, нейтрализуют до pH=8, приливают 1 см3 серной кислоты (1:1) и 0,3 см3 концентрированной фосфорной кислоты, доливают раствор до 100 см3 дистиллированной водой, перемешивают, добавляют 2 см3 раствора дифенилкарбазида с концентрацией 0,5 мас.%, перемешивают и через 5-10 мин фотометрируют с зеленым светофильтром. (ПНДФ 14.1:2.52-96, Методика выполнения измерений массовой концентраций хрома в природных и сточных водах фотометрическим методом с дифенилкарбазидом, М. 1996.) Недостатком способа является его неприемлемость для анализа проб речной воды, содержащей органические и неорганические загрязнители нефтехимических производств (ориентировочный уровень концентраций загрязнителей в исходной анализируемой воде: ХПК до 50 мгО/дм3, pH от 6,0 до 8,5; остальные загрязнители в мг/дм3: взвешенные вещества до 8,0; нефтепродукты до 0,07; сухой остаток до 1000; фенолы до 0,01; аммонийный азот до 1,0; азот нитратный до 1,0; азот нитритный до 0,1; фосфор до 0,2; сульфаты до 300; хлориды до 100; цинк до 0,015; алюминий до 0,1; железо до 0,5; формальдегид до 0,1), анализируемых предварительным концентрированием и минерализацией, в результате чего происходит концентрирование содержащихся примесей до уровня, мешающего данным способом. Сущностью изобретения является обработка сконцентрированной и минерализованной пробы персульфатом аммония и серной кислотой, взятыми в количествах из расчета (0,015-0,02) г и (0,0005 - 0,0006) г моль, соответственно, на каждые 100 см3 исследуемой пробы, взятой на выпаривание, последующая нейтрализация до pH= 8 раствором аммиака или щелочи, фильтрование и определение хрома в фильтрате известным фотометрическим способом по реакции с дифенилкарбазидом. Обработкой пробы персульфатом аммония и серной кислотой, взятыми в количествах прямо пропорциональных объему исследуемой пробы (в отличие от прототипа, где обработку проводят персульфатом аммония и серной кислотой, взятыми в количествах (0,005-0,01) г и 0,0006 г моль, соответственно, независимо от объема пробы), осаждением металлов при pH=8 и последующим фильтрованием (в отличие от прототипа, где фильтрование проводят при кислотности, соответствующей 0,0006 г моль серной кислоты в анализируемом объеме), достигается полное устранение мешающих примесей количественному протеканию реакции хрома с дифенилкарбазидом. При меньшем количестве персульфата, а также отделении осадка при кислотности, соответствующей условиям фильтрования по прототипу, не достигается полное устранение мешающих веществ, присутствующих в пробе, о чем свидетельствует желтая окраска фильтрата и отсутствие малиновой окраски раствора после обработки дифенилкарбазидом или нестабильность образовавшейся окраски во времени. Более высокие количества персульфата и кислоты приводят к неоправданному расходу реактивов и к увеличению продолжительности разложения персульфата, который также мешает дальнейшему определению. Обработка минерализованной и сконцентрированной пробы персульфатом аммония и серной кислотой, взятыми в количествах прямо пропорциональных объему исходной пробы, а также осаждение и фильтрование при pH 8 - являются отличительными признаками и не обнаружены в аналогичных технических решениях, обеспечивают точное определение хрома в речных водах, загрязненных продуктами нефтехимических производств, доступным фотометрическим способом по реакции с дифенилкарбазидом. Наличие отличительных признаков и достигаемого эффекта подтверждают соответствие заявляемого технического решения критериям изобретения: новизна изобретения и изобретательский уровень. Простота анализа, доступность применяемых реактивов и оборудования, обеспечение получения надежных результатов анализа, а также необходимость способа для аналитического контроля речных и сточных вод подтверждают соответствие его критерию "промышленная применимость". Изобретение осуществляется следующим образом. В стакане вместимостью 200-250 см3 выпаривают 250 - 500 см3 исследуемой пробы, в которую предварительно добавляют 0,5 см3 концентрированной азотной и 1,5 см3 концентрированной серной кислот. Выпаривание проводят, приливая пробу в стакан порциями, не превышающими 100 см3. Выпаривание ведут до появления белых паров (примерно до объема 1,5 см3). Если по окончании выпаривания остаток в стакане окрашен в интенсивно желтый цвет или содержит черные крупинки, к содержимому стакана приливают еще 0,5 см3 азотной и 1,0 см3 серной кислот и продолжают до появления белых паров. После охлаждения к минерализованной пробе добавляют, обмывая стенки стакана, 20 см3 дистиллированной воды, нейтрализуют концентрированным раствором аммиака до pH 8 (контролируют по универсальной индикаторной бумаге), приливают 0,6 см3 раствора серной кислоты с концентрацией c(1/2h3SO4)=1,0 моль/дм3 и 0,4 см3 раствора, персульфата аммония с концентрацией 5 мас. % на каждые 100 см3 пробы, упаривают раствор приблизительно до 15 см3, но не менее 30 мин для полного разложения персульфата аммония. Обмывают стенки стакана дистиллированной водой и нейтрализуют раствор до pH = 8 раствором аммиака с концентрацией 10 мас.%. Выпавший осадок отфильтровывают, собирая фильтрат в мерную колбу вместимостью 25 см3, ополаскивают стакан дистиллированной водой, сливая промывные воды в ту же колбу, через тот же фильтр, в колбу вносят 0,3 см3 фосфорной кислоты, разбавленной 1:3, 0,5 см3 серной кислоты, разбавленной 1:3, перемешивают, приливают 1 см3 раствора дифенилкарбазида с концентрацией 0,1 мас.%, перемешивают и через 5-10 мин фотометрируют раствор при длине волны (540
0,3=0,0006 г моль), (5-10) см3 0,1% раствора персульфата аммония (от 0,005 до 0,01 г), кипятят 20-25 мин, фильтруют, упаривают фильтрат до объема 50 см3, нейтрализуют до pH=8, приливают 1 см3 серной кислоты (1:1) и 0,3 см3 концентрированной фосфорной кислоты, доливают раствор до 100 см3 дистиллированной водой, перемешивают, добавляют 2 см3 раствора дифенилкарбазида с концентрацией 0,5 мас.%, перемешивают и через 5-10 мин фотометрируют с зеленым светофильтром. (ПНДФ 14.1:2.52-96, Методика выполнения измерений массовой концентраций хрома в природных и сточных водах фотометрическим методом с дифенилкарбазидом, М. 1996.) Недостатком способа является его неприемлемость для анализа проб речной воды, содержащей органические и неорганические загрязнители нефтехимических производств (ориентировочный уровень концентраций загрязнителей в исходной анализируемой воде: ХПК до 50 мгО/дм3, pH от 6,0 до 8,5; остальные загрязнители в мг/дм3: взвешенные вещества до 8,0; нефтепродукты до 0,07; сухой остаток до 1000; фенолы до 0,01; аммонийный азот до 1,0; азот нитратный до 1,0; азот нитритный до 0,1; фосфор до 0,2; сульфаты до 300; хлориды до 100; цинк до 0,015; алюминий до 0,1; железо до 0,5; формальдегид до 0,1), анализируемых предварительным концентрированием и минерализацией, в результате чего происходит концентрирование содержащихся примесей до уровня, мешающего данным способом. Сущностью изобретения является обработка сконцентрированной и минерализованной пробы персульфатом аммония и серной кислотой, взятыми в количествах из расчета (0,015-0,02) г и (0,0005 - 0,0006) г моль, соответственно, на каждые 100 см3 исследуемой пробы, взятой на выпаривание, последующая нейтрализация до pH= 8 раствором аммиака или щелочи, фильтрование и определение хрома в фильтрате известным фотометрическим способом по реакции с дифенилкарбазидом. Обработкой пробы персульфатом аммония и серной кислотой, взятыми в количествах прямо пропорциональных объему исследуемой пробы (в отличие от прототипа, где обработку проводят персульфатом аммония и серной кислотой, взятыми в количествах (0,005-0,01) г и 0,0006 г моль, соответственно, независимо от объема пробы), осаждением металлов при pH=8 и последующим фильтрованием (в отличие от прототипа, где фильтрование проводят при кислотности, соответствующей 0,0006 г моль серной кислоты в анализируемом объеме), достигается полное устранение мешающих примесей количественному протеканию реакции хрома с дифенилкарбазидом. При меньшем количестве персульфата, а также отделении осадка при кислотности, соответствующей условиям фильтрования по прототипу, не достигается полное устранение мешающих веществ, присутствующих в пробе, о чем свидетельствует желтая окраска фильтрата и отсутствие малиновой окраски раствора после обработки дифенилкарбазидом или нестабильность образовавшейся окраски во времени. Более высокие количества персульфата и кислоты приводят к неоправданному расходу реактивов и к увеличению продолжительности разложения персульфата, который также мешает дальнейшему определению. Обработка минерализованной и сконцентрированной пробы персульфатом аммония и серной кислотой, взятыми в количествах прямо пропорциональных объему исходной пробы, а также осаждение и фильтрование при pH 8 - являются отличительными признаками и не обнаружены в аналогичных технических решениях, обеспечивают точное определение хрома в речных водах, загрязненных продуктами нефтехимических производств, доступным фотометрическим способом по реакции с дифенилкарбазидом. Наличие отличительных признаков и достигаемого эффекта подтверждают соответствие заявляемого технического решения критериям изобретения: новизна изобретения и изобретательский уровень. Простота анализа, доступность применяемых реактивов и оборудования, обеспечение получения надежных результатов анализа, а также необходимость способа для аналитического контроля речных и сточных вод подтверждают соответствие его критерию "промышленная применимость". Изобретение осуществляется следующим образом. В стакане вместимостью 200-250 см3 выпаривают 250 - 500 см3 исследуемой пробы, в которую предварительно добавляют 0,5 см3 концентрированной азотной и 1,5 см3 концентрированной серной кислот. Выпаривание проводят, приливая пробу в стакан порциями, не превышающими 100 см3. Выпаривание ведут до появления белых паров (примерно до объема 1,5 см3). Если по окончании выпаривания остаток в стакане окрашен в интенсивно желтый цвет или содержит черные крупинки, к содержимому стакана приливают еще 0,5 см3 азотной и 1,0 см3 серной кислот и продолжают до появления белых паров. После охлаждения к минерализованной пробе добавляют, обмывая стенки стакана, 20 см3 дистиллированной воды, нейтрализуют концентрированным раствором аммиака до pH 8 (контролируют по универсальной индикаторной бумаге), приливают 0,6 см3 раствора серной кислоты с концентрацией c(1/2h3SO4)=1,0 моль/дм3 и 0,4 см3 раствора, персульфата аммония с концентрацией 5 мас. % на каждые 100 см3 пробы, упаривают раствор приблизительно до 15 см3, но не менее 30 мин для полного разложения персульфата аммония. Обмывают стенки стакана дистиллированной водой и нейтрализуют раствор до pH = 8 раствором аммиака с концентрацией 10 мас.%. Выпавший осадок отфильтровывают, собирая фильтрат в мерную колбу вместимостью 25 см3, ополаскивают стакан дистиллированной водой, сливая промывные воды в ту же колбу, через тот же фильтр, в колбу вносят 0,3 см3 фосфорной кислоты, разбавленной 1:3, 0,5 см3 серной кислоты, разбавленной 1:3, перемешивают, приливают 1 см3 раствора дифенилкарбазида с концентрацией 0,1 мас.%, перемешивают и через 5-10 мин фотометрируют раствор при длине волны (540  15) нм по отношению к дистиллированной воде. В аналогичных условиях проводят контрольный опыт на реактивы, значение оптической плотности которого вычитают из величины оптической плотности рабочего опыта. Калибровочный график строят, включая операцию обработки раствора смесью персульфата аммония и серной кислоты, следующим образом. В термостойкие стаканы вместимостью 100 см3 вносят 0,5-1- 2-3-4-5 см3 стандартного раствор хрома в дистиллированной воде, с концентрацией 0,001 мг/см3, добавляют 40 см3 дистиллированной воды, 1,5 см3 концентрированной серной кислоты, нейтрализуют раствор до pH 8 раствором аммиака с массовой долей 25% (4,0-4,5 см3), приливают 0,8 см3 раствора серной кислоты с молярной концентрацией 0,5 моль/дм3, 0,5 см3 раствора персульфата и кипятят в условиях рабочего опыта при периодическом перемешивании. Затем раствор повторно нейтрализуют до pH 8, количественно переводят в мерную колбу вместимостью 25 см3 и продолжают как при выполнении анализа. Изобретение иллюстрируется следующими примерами. Пример 1. Анализируют пробу реки Тунгуча в условиях прототипа. 1. Построение калибровочного графика. В мерную колбу вместимостью 100 см3 вносят 0,00 - 1,00 -2,00 - 3,00 - 5,00 - 8,00 - 10,00 см3 стандартного раствора хрома с концентрацией 0,001 мг/см3, приливают 1 см3 раствора серной кислоты, разбавленной дистиллированной водой в соотношении 1:1, 0,3 см3 концентрированного раствора фосфорной кислоты и доводят объем до 100 см3 дистиллированной водой, перемешивают, вносят 1,0 см3 1%-ного спиртового раствора дифенилкарбазида и через 10 мин замеряют оптические плотности растворов по отношению к раствору контрольного опыта, который проводят в тех же условиях без стандартного раствора. Получены результаты (в единицах оптической плотности): 0,00: 0,068; 0,137, 0,204; 0,335; 0,520; 0,650, соответственно. 2. Анализ пробы воды. В стакане вместимостью 200-250 см3 выпаривают 500 см3 исследуемой пробы воды, которую предварительно смешивают с 0,5 см3 концентрированной азотной и 1,5 см3 концентрированной серной кислот. Выпаривание проводят, приливая пробу в стакан порциями, не превышающими 100 см3, до появления белых паров (примерно до объема 1,5 см3). После охлаждения к минерализованной пробе добавляют, обмывая стенки стакана, 20 см3 дистиллированной воды, нейтрализуют раствором щелочи с концентрацией 1 моль/дм3, приливают 0,3 см3 раствора серной кислоты с концентрацией 1 моль/дм3, 5-10 см3 раствора персульфата аммония с концентрацией 0,1 мас.% (соответствует 0,005-0,01 сухой соли), кипятят 30 мин, раствор фильтруют, фильтрат выпаривают до 50 см3, охлаждают, переносят в мерную колбу вместимостью 100 см3, нейтрализуют раствором щелочи до pH= 8, приливают 1 см3 раствора серной кислоты 1:1, 0,3 см3 концентрированного раствора фосфорной кислоты и доводят объем до 100 см3 дистиллированной водой, перемешивают, вносят 2,0 см3 1%-ного спиртового раствора дифенилкарбазида и через 10 мин замеряют оптическую плотность раствора. Результат: вместо ожидаемой малиновой окраски наблюдается окрашивание раствора в желтый цвет. Пример 2. Анализируют пробу реки Тунгуча в условиях изобретения. 1. Построение калибровочного графика. В термостойкие стаканы вместимостью 100 см3 отмеривают пипеткой 0,5-1,0-2,0-3,0-4,0-5,0 см3 стандартного раствора с содержанием хрома (VI) 0,001 мг/см3, добавляют 40 см3 дистиллированной воды, 1,5 см3 концентрированной серной кислоты и полученный раствор нейтрализуют до pH=8 раствором щелочи, контролируя значение pH по универсальной индикаторной бумаге, приливают пипеткой 3,0 см3 раствора серной кислоты с молярной концентрацией эквивалента c(1/2h3SO4) = 1 моль/дм3, добавляют 2,0 см3 раствора персульфата с концентрацией 5 мас.% (0,1 г в расчете на сухую соль) кипятят, упаривая раствор приблизительно до 15 см3, но не менее 30 мин, после охлаждения нейтрализуют раствор до pH=8, количественно переносят в мерную колбу вместимостью 25 см3. В колбу вносят 0,25 см3 разбавленного 1:2 фосфорной кислоты, 0,5 см3 разбавленного 1: 2 раствора серной кислоты, перемешивают, добавляют 1 см3 раствора дифенилкарбазида с массовой долей 1%, доливают до метки дистиллированной водой, перемешивают и через 10 мин фотометрируют раствор при зеленом светофильтре по отношению к раствору холостого опыта, который проводят в тех же условиях, но без стандартного раствора. Получены результаты: 0,00; 0,05; 0,11; 0,20; 0,31; 0,43; 0,50, соответственно. По полученным данным строят калибровочный график, откладывая по оси абсцисс количество хрома в фотометрируемом растворе в мг, по оси ординат - соответствующие значения оптических плотностей. 2. Анализ воды. В стакане вместимостью 250 см3 выпаривают 500 см3 исследуемой пробы, в которую предварительно добавляют 0,5 см3 концентрированной азотной и 1,5 см3 концентрированной серной кислот. Выпаривание проводят, приливая пробу в стакан порциями, не превышающими 100 см3. После приливания последней порции пробы выпаривание ведут до объема 40-50 см3, уменьшают температуру нагрева и продолжают упаривание, не допуская разбрызгивания пробы, до появления белых паров серной кислоты (примерно до объема 1,5 см3) Стенки стакана обмывают 5 см3 дистиллированной воды. После охлаждения к минерализованной пробе добавляют, обмывая стенки стакана, 20 см3 дистиллированной воды, перемешивают круговыми движениями, и количественно переносят в термостойкий стакан вместимостью 100 см3. Полученный раствор нейтрализуют до pH = 8, контролируя значение pH по универсальной индикаторной бумаге. Приливают 3 см3 серной кислоты с молярной концентрацией эквивалента c(1/2 h3SO4) = 1 моль/дм3, 2 см3 раствора персульфата аммония с концентрацией 5 мас.% и кипятят, упаривая раствор приблизительно до 15 см3, но не менее 30 мин. Полученный раствор нейтрализуют до pH=8 раствором щелочи, контролируя значение pH по универсальной индикаторной бумаге. Выпавший окрашенный осадок отфильтровывают через фильтр "белая лента", собирая фильтрат в мерную колбу вместимостью 25 см3. Промывают стакан двумя порциями по 3 см3 дистиллированной воды, сливая промывные воды на фильтр с осадком и собирая их в ту же мерную колбу. В колбу вносят 0,25 см3 разбавленного 1: 2 раствора фосфорной кислоты, 0,5 см3 разбавленного 1:2 раствора серной кислоты, перемешивают, добавляют 0,5 см3 раствора дифенилкарбазида с массовой долей 1 мас.%, доливают до метки дистиллированной водой, перемешивают и через 10 мин фотометрируют раствор при зеленом светофильтре (длина волны (540 15) нм) по отношению к дистиллированной воде. В аналогичных условиях проводят контрольный опыт на реактивы, значение оптической плотности которого вычитают из величины оптической плотности рабочего опыта. Результаты анализов: dp - dx = 0,18. Обработка результатов измерений. Количеств хрома, соответствующее значению оптической плотности 0,38, по калибровочному графику = 0,0018 мг. Рассчитывают содержание хрома (Cr) в воде в мг/дм3:
15) нм по отношению к дистиллированной воде. В аналогичных условиях проводят контрольный опыт на реактивы, значение оптической плотности которого вычитают из величины оптической плотности рабочего опыта. Калибровочный график строят, включая операцию обработки раствора смесью персульфата аммония и серной кислоты, следующим образом. В термостойкие стаканы вместимостью 100 см3 вносят 0,5-1- 2-3-4-5 см3 стандартного раствор хрома в дистиллированной воде, с концентрацией 0,001 мг/см3, добавляют 40 см3 дистиллированной воды, 1,5 см3 концентрированной серной кислоты, нейтрализуют раствор до pH 8 раствором аммиака с массовой долей 25% (4,0-4,5 см3), приливают 0,8 см3 раствора серной кислоты с молярной концентрацией 0,5 моль/дм3, 0,5 см3 раствора персульфата и кипятят в условиях рабочего опыта при периодическом перемешивании. Затем раствор повторно нейтрализуют до pH 8, количественно переводят в мерную колбу вместимостью 25 см3 и продолжают как при выполнении анализа. Изобретение иллюстрируется следующими примерами. Пример 1. Анализируют пробу реки Тунгуча в условиях прототипа. 1. Построение калибровочного графика. В мерную колбу вместимостью 100 см3 вносят 0,00 - 1,00 -2,00 - 3,00 - 5,00 - 8,00 - 10,00 см3 стандартного раствора хрома с концентрацией 0,001 мг/см3, приливают 1 см3 раствора серной кислоты, разбавленной дистиллированной водой в соотношении 1:1, 0,3 см3 концентрированного раствора фосфорной кислоты и доводят объем до 100 см3 дистиллированной водой, перемешивают, вносят 1,0 см3 1%-ного спиртового раствора дифенилкарбазида и через 10 мин замеряют оптические плотности растворов по отношению к раствору контрольного опыта, который проводят в тех же условиях без стандартного раствора. Получены результаты (в единицах оптической плотности): 0,00: 0,068; 0,137, 0,204; 0,335; 0,520; 0,650, соответственно. 2. Анализ пробы воды. В стакане вместимостью 200-250 см3 выпаривают 500 см3 исследуемой пробы воды, которую предварительно смешивают с 0,5 см3 концентрированной азотной и 1,5 см3 концентрированной серной кислот. Выпаривание проводят, приливая пробу в стакан порциями, не превышающими 100 см3, до появления белых паров (примерно до объема 1,5 см3). После охлаждения к минерализованной пробе добавляют, обмывая стенки стакана, 20 см3 дистиллированной воды, нейтрализуют раствором щелочи с концентрацией 1 моль/дм3, приливают 0,3 см3 раствора серной кислоты с концентрацией 1 моль/дм3, 5-10 см3 раствора персульфата аммония с концентрацией 0,1 мас.% (соответствует 0,005-0,01 сухой соли), кипятят 30 мин, раствор фильтруют, фильтрат выпаривают до 50 см3, охлаждают, переносят в мерную колбу вместимостью 100 см3, нейтрализуют раствором щелочи до pH= 8, приливают 1 см3 раствора серной кислоты 1:1, 0,3 см3 концентрированного раствора фосфорной кислоты и доводят объем до 100 см3 дистиллированной водой, перемешивают, вносят 2,0 см3 1%-ного спиртового раствора дифенилкарбазида и через 10 мин замеряют оптическую плотность раствора. Результат: вместо ожидаемой малиновой окраски наблюдается окрашивание раствора в желтый цвет. Пример 2. Анализируют пробу реки Тунгуча в условиях изобретения. 1. Построение калибровочного графика. В термостойкие стаканы вместимостью 100 см3 отмеривают пипеткой 0,5-1,0-2,0-3,0-4,0-5,0 см3 стандартного раствора с содержанием хрома (VI) 0,001 мг/см3, добавляют 40 см3 дистиллированной воды, 1,5 см3 концентрированной серной кислоты и полученный раствор нейтрализуют до pH=8 раствором щелочи, контролируя значение pH по универсальной индикаторной бумаге, приливают пипеткой 3,0 см3 раствора серной кислоты с молярной концентрацией эквивалента c(1/2h3SO4) = 1 моль/дм3, добавляют 2,0 см3 раствора персульфата с концентрацией 5 мас.% (0,1 г в расчете на сухую соль) кипятят, упаривая раствор приблизительно до 15 см3, но не менее 30 мин, после охлаждения нейтрализуют раствор до pH=8, количественно переносят в мерную колбу вместимостью 25 см3. В колбу вносят 0,25 см3 разбавленного 1:2 фосфорной кислоты, 0,5 см3 разбавленного 1: 2 раствора серной кислоты, перемешивают, добавляют 1 см3 раствора дифенилкарбазида с массовой долей 1%, доливают до метки дистиллированной водой, перемешивают и через 10 мин фотометрируют раствор при зеленом светофильтре по отношению к раствору холостого опыта, который проводят в тех же условиях, но без стандартного раствора. Получены результаты: 0,00; 0,05; 0,11; 0,20; 0,31; 0,43; 0,50, соответственно. По полученным данным строят калибровочный график, откладывая по оси абсцисс количество хрома в фотометрируемом растворе в мг, по оси ординат - соответствующие значения оптических плотностей. 2. Анализ воды. В стакане вместимостью 250 см3 выпаривают 500 см3 исследуемой пробы, в которую предварительно добавляют 0,5 см3 концентрированной азотной и 1,5 см3 концентрированной серной кислот. Выпаривание проводят, приливая пробу в стакан порциями, не превышающими 100 см3. После приливания последней порции пробы выпаривание ведут до объема 40-50 см3, уменьшают температуру нагрева и продолжают упаривание, не допуская разбрызгивания пробы, до появления белых паров серной кислоты (примерно до объема 1,5 см3) Стенки стакана обмывают 5 см3 дистиллированной воды. После охлаждения к минерализованной пробе добавляют, обмывая стенки стакана, 20 см3 дистиллированной воды, перемешивают круговыми движениями, и количественно переносят в термостойкий стакан вместимостью 100 см3. Полученный раствор нейтрализуют до pH = 8, контролируя значение pH по универсальной индикаторной бумаге. Приливают 3 см3 серной кислоты с молярной концентрацией эквивалента c(1/2 h3SO4) = 1 моль/дм3, 2 см3 раствора персульфата аммония с концентрацией 5 мас.% и кипятят, упаривая раствор приблизительно до 15 см3, но не менее 30 мин. Полученный раствор нейтрализуют до pH=8 раствором щелочи, контролируя значение pH по универсальной индикаторной бумаге. Выпавший окрашенный осадок отфильтровывают через фильтр "белая лента", собирая фильтрат в мерную колбу вместимостью 25 см3. Промывают стакан двумя порциями по 3 см3 дистиллированной воды, сливая промывные воды на фильтр с осадком и собирая их в ту же мерную колбу. В колбу вносят 0,25 см3 разбавленного 1: 2 раствора фосфорной кислоты, 0,5 см3 разбавленного 1:2 раствора серной кислоты, перемешивают, добавляют 0,5 см3 раствора дифенилкарбазида с массовой долей 1 мас.%, доливают до метки дистиллированной водой, перемешивают и через 10 мин фотометрируют раствор при зеленом светофильтре (длина волны (540 15) нм) по отношению к дистиллированной воде. В аналогичных условиях проводят контрольный опыт на реактивы, значение оптической плотности которого вычитают из величины оптической плотности рабочего опыта. Результаты анализов: dp - dx = 0,18. Обработка результатов измерений. Количеств хрома, соответствующее значению оптической плотности 0,38, по калибровочному графику = 0,0018 мг. Рассчитывают содержание хрома (Cr) в воде в мг/дм3: где 500 - объем пробы воды, взятый на анализ, см3; 1000 - коэффициент пересчета в дм3. Пример 3. Анализируют искусственную смесь в соответствии с изобретением (к 1 дм3 воды реки Тунгуча, содержащей 0,005 мг/дм3 хрома по результатам анализа в соответствии с примером 1, добавлено 0,003 мг хрома). Ожидаемая концентрация хрома 0,008 мг/дм3. Анализ проводят в соответствии с примером 2, отбирая на анализ 250 см3 пробы. Результат анализа: 0,0075 мг/дм3,
где 500 - объем пробы воды, взятый на анализ, см3; 1000 - коэффициент пересчета в дм3. Пример 3. Анализируют искусственную смесь в соответствии с изобретением (к 1 дм3 воды реки Тунгуча, содержащей 0,005 мг/дм3 хрома по результатам анализа в соответствии с примером 1, добавлено 0,003 мг хрома). Ожидаемая концентрация хрома 0,008 мг/дм3. Анализ проводят в соответствии с примером 2, отбирая на анализ 250 см3 пробы. Результат анализа: 0,0075 мг/дм3, ФОРМУЛА ИЗОБРЕТЕНИЯ
Способ определения микроколичеств общего хрома в воде, включающий выпаривание пробы и последующую минерализацию, нейтрализацию и обработку минерализованной пробы персульфатом аммония в присутствии серной кислоты, фильтрование, обработку фильтрата дифенилкарбазидом в присутствии серной и фосфорной кислот и фотометрирование окрашенного раствора, отличающийся тем, что обработку персульфатом аммония в присутствии серной кислоты проводят при их количествах, взятых из расчета 0,015-0,02 г 0,0005-0,0006 гмоль соответственно на каждые 100 см3 исследуемой пробы, взятой на выпаривание, а последующее фильтрование проводят после нейтрализации до рН 8. www.freepatent.ru
Способ определения микроколичеств общего хрома в воде
Изобретение относится к области аналитической химии, а именно к способу определения общего хрома (CrIII и CrVI) в воде рыбохозяйственных водоемов с нормой по содержанию хрома не более 0,001 мг/дм3. Способ включает предварительное концентрирование хрома путем выпаривания и обработку, позволяющую устранить мешающее влияние примесей фотометрическому определению хрома по реакции с дифенилкарбазидом.ОПИСАНИЕ ИЗОБРЕТЕНИЯ К ПАТЕНТУ
Изобретение относится к области аналитической химии, а именно к способу определения общего содержания хрома (CrIII и CrVI) в воде рыбохозяйственных водоемов с содержанием хрома в пределах 0,001 до 0,01 мг/дм3. Известен способ определения хрома методом пламенной атомно-абсорбционной спектрометрии (ПНДФ 14.1:2:22-95. Количественный химический анализ вод. Методика выполнения измерений массовой концентрации железа, кадмия, свинца, цинка и хрома в пробах природных и сточных вод методом пламенной атомно-абсорбционной спектрометрии), чувствительность метода 0,02 мг/дм3. Недостатком способа является его недостаточная чувствительность для анализа вод с нормой по содержанию хрома "не более 0,001 мг/дм3", к каким могут быть отнесены пробы воды с показателем pH от 7,0 до 8,5, содержащих восстановители и (или) окрашенные органические вещества, в которых из-за невозможности раздельного определения CrIII и CrVI, определяют только общее содержание хрома, однако, для санитарно-гигиенической оценки качества воды расчеты ведут по наиболее токсичному (ПДК = 0,001 мг/дм3) хрому VI. (Ю.В. Новиков и др., Методы исследования качества воды водоемов, Изд. "Медицина", М. 1990, с. 228). Известен способ фотометрического определения малых количеств суммарного содержания хрома в водах, имеющих щелочную реакцию или содержащих большие количества органических веществ, хлоридов, по которому в пробе, содержащем от 0,001 до 0,05 мг хрома, сначала восстанавливают хром (VI) до хрома (III) сернистой кислотой, затем осаждают хром (III) оксидом магния, отфильтровывают, высушивают, сжигают, растворяют в серной кислоте, окисляют хром (III) до хрома (VI) персульфатом аммония или прокаливанием со смесью карбоната натрия и оксида магния, приливают раствор дифенилкарбазида и фотометрируют. (Ю. Ю. Лурье. Аналитическая химия промышленных сточных вод, Изд. "Химия". М. 1984. с. 155.). Недостатком способа является трудоемкость и высокая погрешность определения, связанная с его многостадийностью, и недостаточная чувствительность (0,02 мг/дм3) для анализа природных вод с нормой по содержанию хрома 0,001 мг/дм3. Известен способ определения хрома в промышленных сточных водах рентгенофлуоресцентным методом (ПНДФ 14.1.43.96. Количественный химический анализ вод. Методика выполнения измерений массовой концентрации ванадия, хрома, марганца, железа, кобальта, никеля, меди, цинка, свинца и висмута в промышленных сточных водах рентгенофлуоресцентным методом), чувствительность метода 0,01 мг/дм3. Недостатком способа является его низкая чувствительность и необходимость дорогостоящего специального оборудования. Известен способ определения общего хрома в воде с помощью атомно-абсорбционной спектрометрии при электротермической атомизации пробы (Г.С. Фомин, Вода. Контроль химической, бактериальной и радиационной безопасности по международным стандартам. Энциклопедический справочник. ИСО 9174, Метод "В",), чувствительность метода 0,005 мг/дм3. Недостатком способа является его неприемлемость для аналитического контроля воды рыбохозяйственных водоемов, для которых предельно допустимая концентрация (ПДК) по содержанию хрома составляет 0,001 мг/дм3. Известен способ определения общего хрома в пробах природной питьевой и сточной воды на анализаторе Флюорат-2, основанный на регистрации свечения хемилюминесценции реакции окисления люминола раствором пероксида водорода, катализируемого солями хрома (III) в щелочной среде (ПНДФ 14.1:2:4. 30-95, Количественный химический анализ вод. Методика выполнения измерений массовой концентрации хрома общего в пробах природной, питьевой и сточной воды на анализаторе жидкости "Флюорат-02"), Минимально определяемая концентрация 0,002 мг/дм3. Недостатком способа является необходимость применения дорогостоящего специального оборудования. Известен способ определения общего хрома в пробах питьевых природных и сточных вод методом инверсионной вольтамперометрии, основанный на адсорбционном концентрировании на поверхности графитового электрода 1,5-дифенилкарбозоната хрома (III), образовавшегося в результате реакции дифенилкарбозона с ионами Cr(VI). Минимально определяемая концентрация хрома 0,001 мг/дм3 (ПНДФ 14.1:2:4.72-96, Количественный химический анализ вод. Методика выполнения измерений массовой концентрации хрома в пробах питьевых, природных и сточных вод методом инверсионной вольтамперометрии.). Недостатком способа является его неприемлемость для анализа вод, содержащих органические загрязнения (содержание органического углерода не должно превышать 0,001 мг/дм3) и необходимость применения специфического оборудования. Наиболее близким по технической сущности и достигаемому результату является способ фотометрического определения общего содержания хрома в природных и сточных водах по реакции хрома (VI) с дифенилкарбазидом, по которому отбирают такой объем исследуемой пробы (разбавленной, сконцентрированной упариванием, минерализованной), в котором содержится (0,001-0,05) мг хрома, доводят объем до 100 см3, нейтрализуют пробу до pH=8 кислотой или щелочью, добавляют 0,3 см3 раствора серной кислоты с концентрацией с(1/2h3SO4)=1 моль/дм3 (1: 2:1000·0,3=0,0006 г · моль), (5-10) см3 0,1% раствора персульфата аммония (от 0,005 до 0,01 г), кипятят 20-25 мин, фильтруют, упаривают фильтрат до объема 50 см3, нейтрализуют до pH=8, приливают 1 см3 серной кислоты (1:1) и 0,3 см3 концентрированной фосфорной кислоты, доливают раствор до 100 см3 дистиллированной водой, перемешивают, добавляют 2 см3 раствора дифенилкарбазида с концентрацией 0,5 мас.%, перемешивают и через 5-10 мин фотометрируют с зеленым светофильтром. (ПНДФ 14.1:2.52-96, Методика выполнения измерений массовой концентраций хрома в природных и сточных водах фотометрическим методом с дифенилкарбазидом, М. 1996.) Недостатком способа является его неприемлемость для анализа проб речной воды, содержащей органические и неорганические загрязнители нефтехимических производств (ориентировочный уровень концентраций загрязнителей в исходной анализируемой воде: ХПК до 50 мгО/дм3, pH от 6,0 до 8,5; остальные загрязнители в мг/дм3: взвешенные вещества до 8,0; нефтепродукты до 0,07; сухой остаток до 1000; фенолы до 0,01; аммонийный азот до 1,0; азот нитратный до 1,0; азот нитритный до 0,1; фосфор до 0,2; сульфаты до 300; хлориды до 100; цинк до 0,015; алюминий до 0,1; железо до 0,5; формальдегид до 0,1), анализируемых предварительным концентрированием и минерализацией, в результате чего происходит концентрирование содержащихся примесей до уровня, мешающего данным способом. Сущностью изобретения является обработка сконцентрированной и минерализованной пробы персульфатом аммония и серной кислотой, взятыми в количествах из расчета (0,015-0,02) г и (0,0005 - 0,0006) г · моль, соответственно, на каждые 100 см3 исследуемой пробы, взятой на выпаривание, последующая нейтрализация до pH= 8 раствором аммиака или щелочи, фильтрование и определение хрома в фильтрате известным фотометрическим способом по реакции с дифенилкарбазидом. Обработкой пробы персульфатом аммония и серной кислотой, взятыми в количествах прямо пропорциональных объему исследуемой пробы (в отличие от прототипа, где обработку проводят персульфатом аммония и серной кислотой, взятыми в количествах (0,005-0,01) г и 0,0006 г · моль, соответственно, независимо от объема пробы), осаждением металлов при pH=8 и последующим фильтрованием (в отличие от прототипа, где фильтрование проводят при кислотности, соответствующей 0,0006 г · моль серной кислоты в анализируемом объеме), достигается полное устранение мешающих примесей количественному протеканию реакции хрома с дифенилкарбазидом. При меньшем количестве персульфата, а также отделении осадка при кислотности, соответствующей условиям фильтрования по прототипу, не достигается полное устранение мешающих веществ, присутствующих в пробе, о чем свидетельствует желтая окраска фильтрата и отсутствие малиновой окраски раствора после обработки дифенилкарбазидом или нестабильность образовавшейся окраски во времени. Более высокие количества персульфата и кислоты приводят к неоправданному расходу реактивов и к увеличению продолжительности разложения персульфата, который также мешает дальнейшему определению. Обработка минерализованной и сконцентрированной пробы персульфатом аммония и серной кислотой, взятыми в количествах прямо пропорциональных объему исходной пробы, а также осаждение и фильтрование при pH 8 - являются отличительными признаками и не обнаружены в аналогичных технических решениях, обеспечивают точное определение хрома в речных водах, загрязненных продуктами нефтехимических производств, доступным фотометрическим способом по реакции с дифенилкарбазидом. Наличие отличительных признаков и достигаемого эффекта подтверждают соответствие заявляемого технического решения критериям изобретения: новизна изобретения и изобретательский уровень. Простота анализа, доступность применяемых реактивов и оборудования, обеспечение получения надежных результатов анализа, а также необходимость способа для аналитического контроля речных и сточных вод подтверждают соответствие его критерию "промышленная применимость". Изобретение осуществляется следующим образом. В стакане вместимостью 200-250 см3 выпаривают 250 - 500 см3 исследуемой пробы, в которую предварительно добавляют 0,5 см3 концентрированной азотной и 1,5 см3 концентрированной серной кислот. Выпаривание проводят, приливая пробу в стакан порциями, не превышающими 100 см3. Выпаривание ведут до появления белых паров (примерно до объема 1,5 см3). Если по окончании выпаривания остаток в стакане окрашен в интенсивно желтый цвет или содержит черные крупинки, к содержимому стакана приливают еще 0,5 см3 азотной и 1,0 см3 серной кислот и продолжают до появления белых паров. После охлаждения к минерализованной пробе добавляют, обмывая стенки стакана, 20 см3 дистиллированной воды, нейтрализуют концентрированным раствором аммиака до pH 8 (контролируют по универсальной индикаторной бумаге), приливают 0,6 см3 раствора серной кислоты с концентрацией c(1/2h3SO4)=1,0 моль/дм3 и 0,4 см3 раствора, персульфата аммония с концентрацией 5 мас. % на каждые 100 см3 пробы, упаривают раствор приблизительно до 15 см3, но не менее 30 мин для полного разложения персульфата аммония. Обмывают стенки стакана дистиллированной водой и нейтрализуют раствор до pH = 8 раствором аммиака с концентрацией 10 мас.%. Выпавший осадок отфильтровывают, собирая фильтрат в мерную колбу вместимостью 25 см3, ополаскивают стакан дистиллированной водой, сливая промывные воды в ту же колбу, через тот же фильтр, в колбу вносят 0,3 см3 фосфорной кислоты, разбавленной 1:3, 0,5 см3 серной кислоты, разбавленной 1:3, перемешивают, приливают 1 см3 раствора дифенилкарбазида с концентрацией 0,1 мас.%, перемешивают и через 5-10 мин фотометрируют раствор при длине волны (540 ± 15) нм по отношению к дистиллированной воде. В аналогичных условиях проводят контрольный опыт на реактивы, значение оптической плотности которого вычитают из величины оптической плотности рабочего опыта. Калибровочный график строят, включая операцию обработки раствора смесью персульфата аммония и серной кислоты, следующим образом. В термостойкие стаканы вместимостью 100 см3 вносят 0,5-1- 2-3-4-5 см3 стандартного раствор хрома в дистиллированной воде, с концентрацией 0,001 мг/см3, добавляют 40 см3 дистиллированной воды, 1,5 см3 концентрированной серной кислоты, нейтрализуют раствор до pH 8 раствором аммиака с массовой долей 25% (4,0-4,5 см3), приливают 0,8 см3 раствора серной кислоты с молярной концентрацией 0,5 моль/дм3, 0,5 см3 раствора персульфата и кипятят в условиях рабочего опыта при периодическом перемешивании. Затем раствор повторно нейтрализуют до pH 8, количественно переводят в мерную колбу вместимостью 25 см3 и продолжают как при выполнении анализа. Изобретение иллюстрируется следующими примерами. Пример 1. Анализируют пробу реки Тунгуча в условиях прототипа. 1. Построение калибровочного графика. В мерную колбу вместимостью 100 см3 вносят 0,00 - 1,00 -2,00 - 3,00 - 5,00 - 8,00 - 10,00 см3 стандартного раствора хрома с концентрацией 0,001 мг/см3, приливают 1 см3 раствора серной кислоты, разбавленной дистиллированной водой в соотношении 1:1, 0,3 см3 концентрированного раствора фосфорной кислоты и доводят объем до 100 см3 дистиллированной водой, перемешивают, вносят 1,0 см3 1%-ного спиртового раствора дифенилкарбазида и через 10 мин замеряют оптические плотности растворов по отношению к раствору контрольного опыта, который проводят в тех же условиях без стандартного раствора. Получены результаты (в единицах оптической плотности): 0,00: 0,068; 0,137, 0,204; 0,335; 0,520; 0,650, соответственно. 2. Анализ пробы воды. В стакане вместимостью 200-250 см3 выпаривают 500 см3 исследуемой пробы воды, которую предварительно смешивают с 0,5 см3 концентрированной азотной и 1,5 см3 концентрированной серной кислот. Выпаривание проводят, приливая пробу в стакан порциями, не превышающими 100 см3, до появления белых паров (примерно до объема 1,5 см3). После охлаждения к минерализованной пробе добавляют, обмывая стенки стакана, 20 см3 дистиллированной воды, нейтрализуют раствором щелочи с концентрацией 1 моль/дм3, приливают 0,3 см3 раствора серной кислоты с концентрацией 1 моль/дм3, 5-10 см3 раствора персульфата аммония с концентрацией 0,1 мас.% (соответствует 0,005-0,01 сухой соли), кипятят 30 мин, раствор фильтруют, фильтрат выпаривают до 50 см3, охлаждают, переносят в мерную колбу вместимостью 100 см3, нейтрализуют раствором щелочи до pH= 8, приливают 1 см3 раствора серной кислоты 1:1, 0,3 см3 концентрированного раствора фосфорной кислоты и доводят объем до 100 см3 дистиллированной водой, перемешивают, вносят 2,0 см3 1%-ного спиртового раствора дифенилкарбазида и через 10 мин замеряют оптическую плотность раствора. Результат: вместо ожидаемой малиновой окраски наблюдается окрашивание раствора в желтый цвет. Пример 2. Анализируют пробу реки Тунгуча в условиях изобретения. 1. Построение калибровочного графика. В термостойкие стаканы вместимостью 100 см3 отмеривают пипеткой 0,5-1,0-2,0-3,0-4,0-5,0 см3 стандартного раствора с содержанием хрома (VI) 0,001 мг/см3, добавляют 40 см3 дистиллированной воды, 1,5 см3 концентрированной серной кислоты и полученный раствор нейтрализуют до pH=8 раствором щелочи, контролируя значение pH по универсальной индикаторной бумаге, приливают пипеткой 3,0 см3 раствора серной кислоты с молярной концентрацией эквивалента c(1/2h3SO4) = 1 моль/дм3, добавляют 2,0 см3 раствора персульфата с концентрацией 5 мас.% (0,1 г в расчете на сухую соль) кипятят, упаривая раствор приблизительно до 15 см3, но не менее 30 мин, после охлаждения нейтрализуют раствор до pH=8, количественно переносят в мерную колбу вместимостью 25 см3. В колбу вносят 0,25 см3 разбавленного 1:2 фосфорной кислоты, 0,5 см3 разбавленного 1: 2 раствора серной кислоты, перемешивают, добавляют 1 см3 раствора дифенилкарбазида с массовой долей 1%, доливают до метки дистиллированной водой, перемешивают и через 10 мин фотометрируют раствор при зеленом светофильтре по отношению к раствору холостого опыта, который проводят в тех же условиях, но без стандартного раствора. Получены результаты: 0,00; 0,05; 0,11; 0,20; 0,31; 0,43; 0,50, соответственно. По полученным данным строят калибровочный график, откладывая по оси абсцисс количество хрома в фотометрируемом растворе в мг, по оси ординат - соответствующие значения оптических плотностей. 2. Анализ воды. В стакане вместимостью 250 см3 выпаривают 500 см3 исследуемой пробы, в которую предварительно добавляют 0,5 см3 концентрированной азотной и 1,5 см3 концентрированной серной кислот. Выпаривание проводят, приливая пробу в стакан порциями, не превышающими 100 см3. После приливания последней порции пробы выпаривание ведут до объема 40-50 см3, уменьшают температуру нагрева и продолжают упаривание, не допуская разбрызгивания пробы, до появления белых паров серной кислоты (примерно до объема 1,5 см3) Стенки стакана обмывают 5 см3 дистиллированной воды. После охлаждения к минерализованной пробе добавляют, обмывая стенки стакана, 20 см3 дистиллированной воды, перемешивают круговыми движениями, и количественно переносят в термостойкий стакан вместимостью 100 см3. Полученный раствор нейтрализуют до pH = 8, контролируя значение pH по универсальной индикаторной бумаге. Приливают 3 см3 серной кислоты с молярной концентрацией эквивалента c(1/2 h3SO4) = 1 моль/дм3, 2 см3 раствора персульфата аммония с концентрацией 5 мас.% и кипятят, упаривая раствор приблизительно до 15 см3, но не менее 30 мин. Полученный раствор нейтрализуют до pH=8 раствором щелочи, контролируя значение pH по универсальной индикаторной бумаге. Выпавший окрашенный осадок отфильтровывают через фильтр "белая лента", собирая фильтрат в мерную колбу вместимостью 25 см3. Промывают стакан двумя порциями по 3 см3 дистиллированной воды, сливая промывные воды на фильтр с осадком и собирая их в ту же мерную колбу. В колбу вносят 0,25 см3 разбавленного 1: 2 раствора фосфорной кислоты, 0,5 см3 разбавленного 1:2 раствора серной кислоты, перемешивают, добавляют 0,5 см3 раствора дифенилкарбазида с массовой долей 1 мас.%, доливают до метки дистиллированной водой, перемешивают и через 10 мин фотометрируют раствор при зеленом светофильтре (длина волны (540 ± 15) нм) по отношению к дистиллированной воде. В аналогичных условиях проводят контрольный опыт на реактивы, значение оптической плотности которого вычитают из величины оптической плотности рабочего опыта. Результаты анализов: dp - dx = 0,18. Обработка результатов измерений. Количеств хрома, соответствующее значению оптической плотности 0,38, по калибровочному графику = 0,0018 мг. Рассчитывают содержание хрома (Cr) в воде в мг/дм3:где 500 - объем пробы воды, взятый на анализ, см3;1000 - коэффициент пересчета в дм3.
ФОРМУЛА ИЗОБРЕТЕНИЯ
Способ определения микроколичеств общего хрома в воде, включающий выпаривание пробы и последующую минерализацию, нейтрализацию и обработку минерализованной пробы персульфатом аммония в присутствии серной кислоты, фильтрование, обработку фильтрата дифенилкарбазидом в присутствии серной и фосфорной кислот и фотометрирование окрашенного раствора, отличающийся тем, что обработку персульфатом аммония в присутствии серной кислоты проводят при их количествах, взятых из расчета 0,015-0,02 г 0,0005-0,0006 г·моль соответственно на каждые 100 см3 исследуемой пробы, взятой на выпаривание, а последующее фильтрование проводят после нейтрализации до рН 8.bankpatentov.ru